BPP assumptions models and input files - bpp/bpp-tutorial-geneflow GitHub Wiki
To ensure this assumption is met a good strategy is to target short (e.g., 500 to 1000 bp) genomic segments (loci).
Note that simulations based results suggests that phylogenomic inferences under the multi-species coalescent model are robust to realistic amounts of intralocus recombination Zhu et al, 2021.
The different loci should be physically distant from one another in the genome. This ensures that recombination between them is common allowing the loci to have approximately independent histories.
Loci should be evolving neutrally, implying that their gene trees are not affected to a significant extent by natural selection
Despite this assumption, protein-coding genes appear to be useable for BPP analysis even if they show obvious evidence of purifying selection. Most proteins perform similar functions in closely related species and the main effect of purifying selection on nonsynonymous mutations is a reduction of the neutral mutation rate for the locus. Studies comparing species trees inferred using exons and using introns or noncoding DNA gave highly consistent results between the two kinds of data (Shi and Yang, 2018). Nevertheless, it is prudent to analyze non-coding and coding regions of the genome as separate data sets.
By default BPP assumes the Jukes-Cantor mutation/substitution model with a constant rate over time or lineages (the molecular clock). These assumptions are reasonable for analysing closely related species with sequence divergences below ∼10%, say. Note that the role of the mutation model in bpp analysis is to correct for multiple hits at the same site, so that the choice of the model (JC versus GTR, say) is unimportant (Shi and Yang 2018). However for distantly related species the clock may be violated, and the model violation may cause incorrect inference. BPP implements more general models such as GTR, and relaxed clocks to allow the rate to vary among species (Flouri et al. 2022). However, the algorithms are found to have mixing problems and can be applied to small datasets only (100 or 500 loci, say). In summary, BPP is currently most suitable for closely related species. To apply BPP to distantly related species, one should take special care to confirm convergence of the runs.
Below is a simple model of MSC depicting a species tree of three species with a gene tree of six sequences evolving inside it. The model consists of seven parameters: two τs (species divergence times), and five θs (population size parameters).
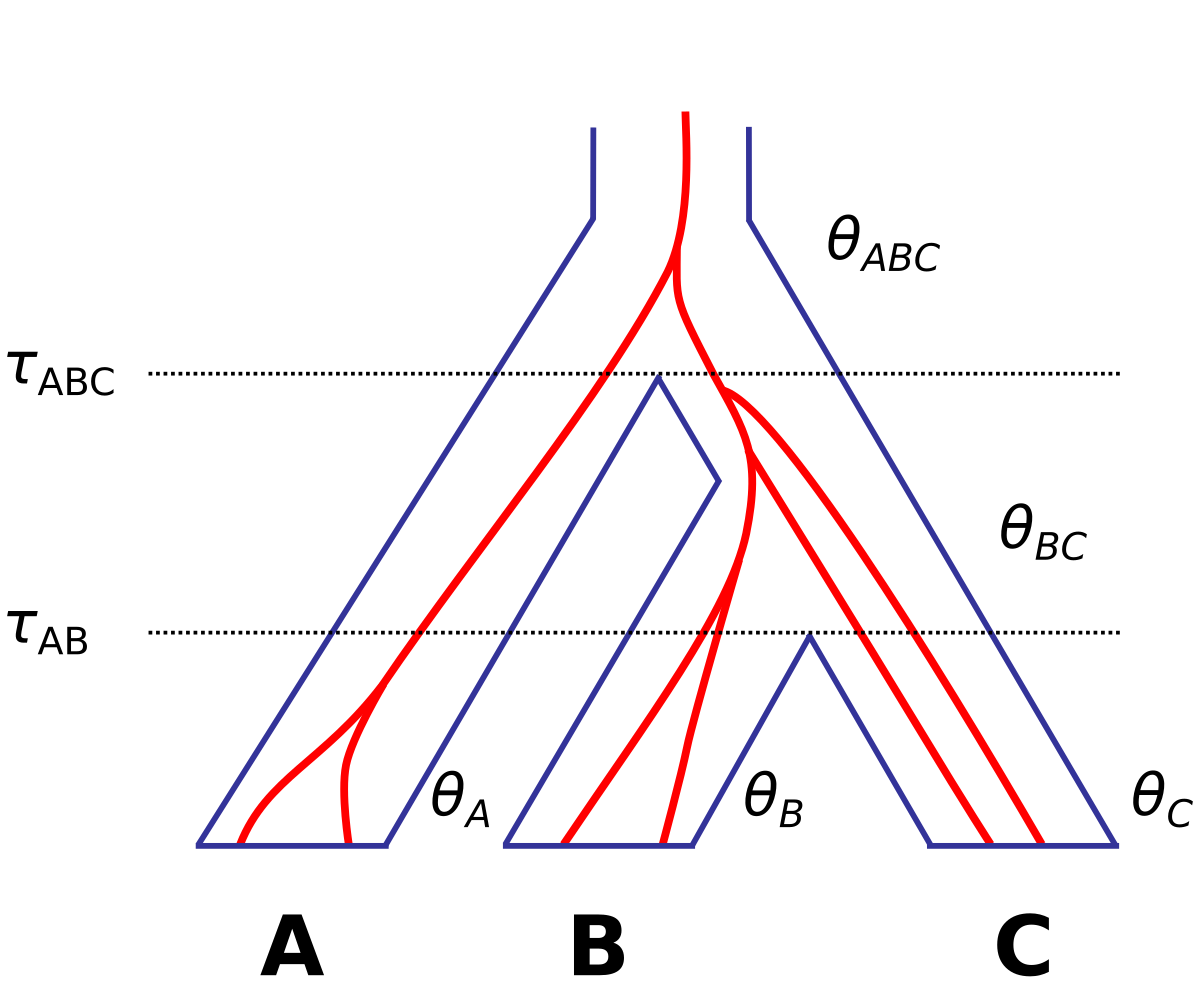
A population size parameter θ=4Nμ is the average proportion of different sites between two sequences sampled at random from the population. N is the effective population size and μ is the mutation rate per site per generation.
Both τ and θ are measured by the expected number of substitutions per site.
In general, if there are s species in the species tree, the model will involve (s-1) species divergence times (τs) and (s-1) ancestral θs. If a species has at least two sequences at any loci, a θ for that species will be used as well. If there is one species, the model will involve one parameter only (θ for that species).
BPP implements four methods for analyzing multi-locus sequence data. They can be specified by setting the control file options speciesdelimitation and speciestree to either 0 (disable) or 1 (enable).
| speciesdelimitation | speciestree | |
| 0 | 1 | |
| 0 | A00. Estimation of parameters under the multispecies coalescent model with or without gene flow (Yang and Rannala, 2003; Flouri et al. 2020; Flouri et al. 2023) | A01. Inference of species tree when the assignment and delimitation are given (Rannala and Yang, 2017; Flouri et al. 2018) |
| 1 | A10. Species delimitation using a fixed guide tree (Yang and Rannala, 2010; Rannala and Yang, 2013) | A11. Joint species delimitation and species-tree inference or unguided species delimitation (Yang and Rannala, 2014) |
In this tutorial we will be working with methods A00 and A01. In the first part of the tutorial, we will estimate a species tree (without accounting for gene flow) for our dataset (method A01). Once we have a species tree, we will estimate the parameters under the multispecies coalescent model again without accounting for geneflow (method A00).
The second part of the tutorial will deal with estimating the parameters of the model in the presence of gene flow.
Note: A species delimitation tutorial is also available here
For all BPP analyses there are three necessary input files:
-
The control file that contains all the parameters for the analysis and the paths to input and output files
-
The Sequence file that contains the aligned sequences
-
The Imap file which contains the information about the assignment of samples to species
Species Tree Inference with Astral | BPP assumptions | BPP control file | Species Tree Inference with BPP | Parameter Estimation with BPP
-
Flouri T., Jiao X., Rannala B., Yang Z. (2018) Species Tree Inference with BPP using Genomic Sequences and the Multispecies Coalescent. Molecular Biology and Evolution, 35(10):2585-2593. doi:10.1093/molbev/msy147
-
Flouri T., Jiao X., Rannala B., Yang Z. (2020) A Bayesian Implementation of the Multispecies Coalescent Model with Introgression for Phylogenomic Analysis. Molecular Biology and Evolution, 37(4):1211-1223. doi:10.1093/molbev/msz296
-
Flouri T., Huang J., Jiao X., Kapli P., Rannala B., Yang Z. (2022) Bayesian Phylogenetic Inference using Relaxed-clocks and the Multispecies Coalescent. Molecular Biology and Evolution, 39(8). doi:10.1093/molbev/msac161
-
Flouri T., Jiao X., Huang J., Rannala B., Yang Z. (2023) Efficient Bayesian inference under the multispecies coalescent with migration. Proceedings of the National Academy of Sciences, 120(44):e2310708120. doi:10.1073/pnas.2310708120
-
Rannala B., Yang Z. (2013) Improved reversible jump algorithms for Bayesian species delimitation. Genetics, 194:245-253. doi:10.1534/genetics.112.149039
-
Rannala B., Yang Z. (2017) Efficient Bayesian Species Tree Inference under the Multispecies Coalescent. Systematic Biology, 66(5):823-842. doi:10.1093/sysbio/syw119
-
Yang Z., Rannala B. (2003) Bayes Estimation of Species Divergence Times and Ancestral Population Sizes using DNA Sequences From Multiple Loci. Genetics, 164:1645-1656. doi:10.1093/genetics/164.4.1645
-
Yang Z., Rannala B. (2010) Bayesian species delimitation using multilocus sequence data. Proceedings of the National Academy of Sciences, 107(20):9264-9269. doi:10.1073/pnas.0913022107
-
Yang Z., Rannala B. (2014) Unguided species delimitation using DNA sequence data from multiple loci. Molecular Biology and Evolution, 31(12):3125-3135. doi:10.1093/molbev/msu279