GunnsFluidMultiAdsorber - nasa/gunns GitHub Wiki

This link is similar to GunnsFluidAdsorber and GunnsFluidHotAdsorber but with several additional features. Refer to those links for more background info. All of these additional features are optional and can be configured:
- It can act on multiple fluid compounds. These can be network fluid types and trace compounds, and all of their absorption & desorption can act independently. Some interaction between them is also supported.
- Sorption rates can taper off to zero when approaching maximum absorbed mass or zero absorbed mass limits. This allows a gradual 'break-through' of the absorbed compound as the adsorber fills up, or a more abrupt break-through right when the absorbed mass limit is reached.
- Interaction of absorbed compounds on on each other's sorbtion rates. This allows things like a Metox CO2 adsorber, which must first absorb some H2O before it can effectively absorb CO2.
- Switches automatically between adsorption/desorption based on pressure and/or temperature.
- Models heat of the sorbing reaction. This can be used to interact with a thermal aspect representing the average temperature of the sorbant mass, which can feed back into the sorption rates.
- Consistent configuration between network fluid types and trace compounds.
The features of GunnsFluidAdsorber and GunnsFluidHotAdsorber that this doesn't keep are:
- There is not a maximum adsorption rate.
- There is not a desorption cycle input flag to control switching between adsorption/desorption directions from outside, as this is controlled automatically from within the model.
All other features of GunnsFluidHotAdsorber, and by extension GunnsFluidAdsorber, are present in this model.
Configuration of this link involves the Trick input file as well as the traditional GunnShow shape data. Each compund must be configured & added to the link via a call from the input file. This is described below.
This link can be connected to nodes in the same ways as a GunnsFluidConductor.
Port Connection Rules (These are limitations on the port connection to nodes that the link enforces in run-time):
- Same as GunnsFluidConductor.
- This link cannot act on liquid types and therefore connection to liquid nodes is not allowed. Ports may ony be connected to Gas nodes or the Ground node.
- The link will throw errors if both ports are connected to the network Ground node during init. After init however, both ports can be moved to Ground.
Other Rules (These are extra rules you should always try to follow):
- N/A.
Configuration Data Parameters:
- maxConductivity: Same as GunnsFluidConductor.
- expansionScaleFactor: Same as GunnsFluidConductor.
- thermalLength: Same as GunnsFluidValve.
- thermalDiameter: Same as GunnsFluidValve.
- surfaceRoughness: Same as GunnsFluidValve.
Input Data Parameters:
- malfBlockageFlag: Same as GunnsFluidConductor.
- malfBlockageValue: Same as GunnsFluidConductor.
- wallTemperature: Same as GunnsFluidValve. This is also the temperature of the sorbant medium.
Input File:
Whereas the GunnShow configuration & input shape data configures the bulk link properties described above, all data pertinent to actual compounds is set via the Trick Input Processor. The link was designed this way because it supports a variable number of compounds, but GunnShow doesn't have enough support for for such variable-sized shape data.
In the simulation's Trick input file, for each compound that the link will sorb, we make one call to the addCompound method in the link's configuration data object in the network. The compound's configuration is provided as arguments to the method.
The syntax for this call in the input file is:
sim_object.other_pathing.network.netConfig.the_link.addCompound(args)
where args are the arguments described below, the bold text is exactly as shown and the non-bold text are the names of your particular sim object, network and link, etc.
The arguments are listed below in the order they are provided:
- Chemical Compound Type (no default, a value must be provided): The chemical compound to be sorbed that is configured with this method call. This is a value of the ChemicalCompound::Type enumeration type listed in ms-utils/properties/ChemicalCompound.hh. For example to add CO2, this field would be trick.ChemicalCompound.CO2
- Maximum Adsorbed Mass (no default, a value > 0 (kg) must be provided): This is the same as maxAdsorbedMass in GunnsFluidAdsorber. This is the maximum mass of this compound that the absorber can absorb.
- Efficiency Bias (no default, a value must be provided): This is the same as efficiencyBias in GunnsFluidHotAdsorber. Just like GunnsFluidHotAdsorber, the sorption efficiency is a linear function of temperature, and this value is the intercept of that line on the efficiency axis, T = 0 K. For models where you want adsorption and desorption efficiency to vary with temperature, then configure this line such that it crosses the zero efficiency axis at the temperature where the adsorber would switch between adsorption & desorption, with the positive region for adsorption and negative region for desorption.
- Efficiency Scale Factor (default = 0 (1/K)): This is the same as efficiencyScaleFactor in GunnsFluidHotAdsorber. This is the slope of the efficiency vs. temperature line described above.
- Desorb Partial Pressure (default = 0 (kPa)): This defines the partial-pressure value of the compound in the gas stream above which the link can absorb the compound, and below which the link wil desorb it. This is used for pressure swing bed adsorbers that tend to absorb the compound at high pressure and desorb it when exposed to vacuum. Note that this link can only absorb when the actual compound partial-pressure in the gas stream is higher than this value. If you don't need to model pressure swinging between absorption & desorption, then leave this value zero.
- Desorb Rate Factor (default = 0 (kg/s/kPa)): This controls how fast the compound will desorb back into the gas stream, in (kg/s) mass rate per (kPa) of difference between the actual compound's partial-pressure in the gas stream, and the Desorb Partial Pressure value set above.
- Heat Of Adsorption (default = 0 (kJ/mol)): This is the heat of reaction (change in enthalpy) of the chemical reaction in the adsorbing direction, per mol of this compound adsorbed. For an exothermic reaction, which most adsorbing reactions are, this will have a negative value. For example in the equation for a dilithium peroxide CO2 adsorption:
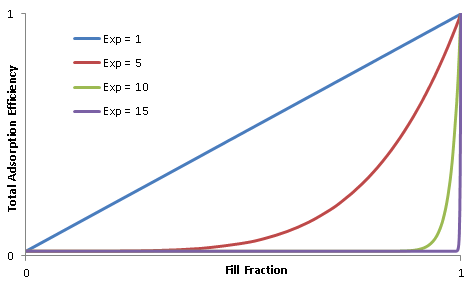
- Taper-Off Flag (default = False): When False, the sorption rate doesn't slow down as the adsorbed mass approaches a limit (max adsorbed mass for absorbing, and zero for desorbing). When True, the sorption rate slows to zero as the adsorbed mass approaches the limit, and the adsorbed mass can never actually reach the exact limit. This models 'breakthrough' and escape of the sorbate through the adsorber as the adsorber's sorbant fills up and loses efficiency. Note that in GunnsFluidAdsorber, its default behavior is False for network fluid constituents, and True for trace compounds. Our flag allows this to be set for each compound regardless of its type. The suddenness at which the breakthrough occurs is configured by the Breakthrough Exponent argument below.
- Dependent Compound Type (default = NO_COMPOUND): this is the chemical compound whose presence in the adsorber affects the sorbing rate of this compound. This uses the same syntax as Chemical Compound Type above. For instance if this compound is CO2 and we want it to be affected by the amount of H2O in the adsorber, then we'd use trick.ChemicalCompound.H2O here. Also note that the dependent compound must also be added to the link in its own call to addCompound. The calling order of these compounds doesn't matter.
- Initial Malfunction Efficiency Flag (default = False): This is the intial activate state of the sorbing efficiency override malfunction for this compound.
- Initial Malfunction Efficiency Value (default = 0, must be (0-1)): This is the initial value for the sorbing efficiency override malfunction for this compound. This scales the adsorption & desorption rates by this value, so a 1 has no affect, and 0 stops sorption entirely.
- Initial Adsorbed Mass (default = 0 (kg), must be >= 0 and <= Maximum Adsorbed Mass): This is the initial mass of this compound already present in the adsorber.
- Breakthrough Exponent (default = 1, must be >= 1 and <= 100): This sets the curvature of the breakthrough effect when the Taper-Off Flag is used. The trend of varying this value is illustrated below. Higher values produce a more sudden breakthrough effect.
the heat is also written as delta-Enthalpy dH and note in this form it is negative:
but note that there are 2 mols of CO2 here, so the actual value we would use is -379.8 / 2 = -189.9 kJ/mol.
Here's an example call adding CO2 and H2O to a link called co2Bed in a network called ars. In this example the CO2 sorption depends on the amount of H2O in the adsorber:
sim_object.ars.netConfig.co2Bed.addCompound(trick.ChemicalCompound.CO2, 1.0, 0.85, 0.0, 0.05, 0.01, -100.0, False, trick.ChemicalCompound.H2O)
sim_object.ars.netConfig.co2Bed.addCompound(trick.ChemicalCompound.H2O, 0.5, 0.75, 0.0, 0.05, 0.01, -87.0, True, trick.ChemicalCompound.NO_COMPOUND, False, 0.0, 0.5)
- N/A
- N/A