Bedtools - lmigueel/Bioinformatica GitHub Wiki
O formato BED é uma maneira concisa e flexível de representar características e anotações genômicas. A descrição do formato BED suporta até 12 colunas, mas apenas as 3 primeiras são necessárias.
bedtools permite usar o formato “BED12” (ou seja, todos os 12 campos listados abaixo). No entanto, apenas intersectBed, coberturaBed, genomeCoverageBed e bamToBed obedecerão aos “blocos” BED12 ao computar sobreposições, etc., por meio da opção “-split”. Para todas as outras ferramentas, as últimas seis colunas não são usadas para nenhuma comparação pelas ferramenta. Em vez disso, eles usarão toda a extensão (do início ao fim) da entrada BED12 para realizar quaisquer comparações de recursos relevantes. As últimas seis colunas serão relatadas na saída de todas as comparações.
chrom - The name of the chromosome on which the genome feature exists.
For example, “chr1”, “contig1112.23”. This column is required.
start - The zero-based starting position of the feature in the chromosome.
The first base in a chromosome is numbered 0. This column is required.
end - The one-based ending position of the feature in the chromosome.
This column is required.
name - Defines the name of the BED feature. This column is optional.
For example, “LINE”, “Exon3”.
score - The UCSC definition requires that a BED score range from 0 to 1000, inclusive.
This column is optional.
strand - Defines the strand - either ‘+’ or ‘-‘. This column is optional.
thickStart - The starting position at which the feature is drawn thickly.
Allowed yet ignored by bedtools.
thickEnd - The ending position at which the feature is drawn thickly.
Allowed yet ignored by bedtools.
itemRgb - An RGB value of the form R,G,B (e.g. 255,0,0).
Allowed yet ignored by bedtools.
blockCount - The number of blocks (exons) in the BED line.
Allowed yet ignored by bedtools.
blockSizes - A comma-separated list of the block sizes.
Allowed yet ignored by bedtools.
blockStarts - A comma-separated list of block starts.
Allowed yet ignored by bedtools.
Eu particularmente utilizo apenas as três primeiras colunas para realizar interseções, junções e afins entre resultados de posições genômicas.
AQUI você acessa a o manual de uso do bedtools. Existem muitos exemplos, mas vamos citar alguns.
bedtools ainda permite cruzar, mesclar, contar, complementar e embaralhar intervalos genômicos de vários arquivos em formatos de arquivo genômico amplamente usados, como BAM, BED, GFF/GTF e VCF.
Via conda:
conda install -c bioconda bedtools Classifica um arquivo por cromossomo e/ou outros critérios do bedtools sort. Por padrão, classifica um arquivo BED por cromossomo e, em seguida, pela posição inicial em ordem crescente.
cat A.bed
chr1 800 1000
chr1 80 180
chr1 1 10
chr1 750 10000
sortBed -i A.bed
chr1 1 10
chr1 80 180
chr1 750 10000
chr1 800 1000
Linha de comando: bedtools sort -i <file.bed> > <output_file.sort.bed>, onde a flag -i significada a entrada no formato BED.
Extrai sequências de um arquivo FASTA para cada um dos intervalos definidos em um arquivo BED/GFF/VCF. Por padrão, o cabeçalho FASTA para cada sequência extraída será formatado da seguinte maneira: “: <início> - ”.

Vamos ao exemplo:
$ cat test.fa
>chr1
AAAAAAAACCCCCCCCCCCCCGCTACTGGGGGGGGGGGGGGGGGG
$ cat test.bed
chr1 5 10
$ bedtools getfasta -fi test.fa -bed test.bed
>chr1:5-10
AAACC
# optionally write to an output file
$ bedtools getfasta -fi test.fa -bed test.bed -fo test.fa.out
$ cat test.fa.out
>chr1:5-10
AAACC
Linha de comando: bedtools getfasta -fi <file.fasta> -bed <file.gff> -s -fo <output_file.fasta>, onde:
-fi: Entrada no formato FASTA.
-bed: Arquivo no formato BED que se deseja extrair as coordenadas.
-s: Considerar a fita. Padrão: as informações da fita são ignoradas.
-fo: Saída no formato FASTA.
Retorna todos os intervalos em um genoma que não são cobertos por pelo menos um intervalo no arquivo de entrada BED/GFF/VCF e assim gera os arquivos com as regiões intergênicas.
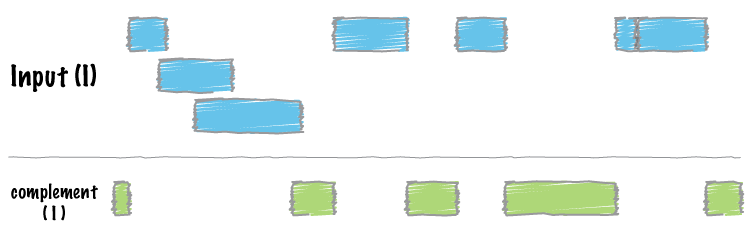
Vamos ao exemplo:
$ cat A.bed
chr1 100 200
chr1 400 500
chr1 500 800
$ cat my.genome
chr1 1000
chr2 800
$ bedtools complement -i A.bed -g my.genome
chr1 0 100
chr1 200 400
chr1 800 1000
chr2 0 800
Linha de comando: bedtools complement -i <file.gff> -g <lenght.txt> > <output_file.itt>, onde
-i: Entrada no formato BED/GFF/VCF.
-g: Arquivo contendo a quantidade de sequência de cada header (exemplo: chr1\t593)
Combina regiões sobrepostas ou com intervalos determinados pelo usuário.
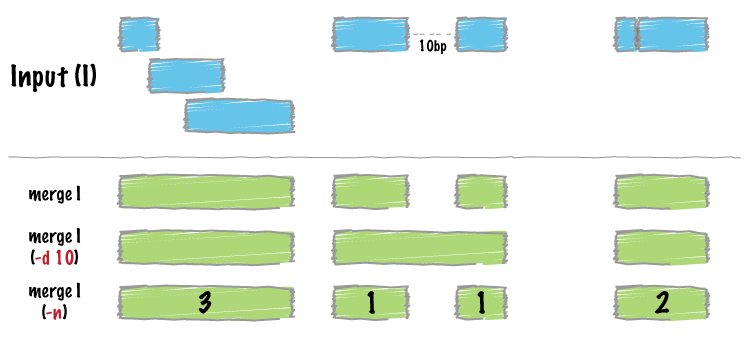
Vamos ao exemplo:
$ cat A.bed
chr1 100 200
chr1 180 250
chr1 250 500
chr1 501 1000
$ bedtools merge -i A.bed
chr1 100 500
chr1 501 1000
Linha de comando: bedtools merge -i <file.bed> -s -c 2,4,4,4,5,6 -o count,mean,min,max,collapse,distinct > <output_file.bed>, onde:
-i: Entrada no formato BED/GFF/VCF.
-s: Considerar a fita. Padrão: as informações da fita são ignoradas.
-c: Especificar as colunas do arquivo de entrada para operar (consulte a opção -o). Várias colunas podem ser especificadas em uma lista delimitada por vírgulas.
-o: Especifique a operação que deve ser aplicada -c. Operações válidas: soma; mínimo; máximo; absmin; absma;, média mediana; recolher (ou seja, imprimir uma lista delimitada (duplicatas permitidas)); distinto (ou seja, imprime uma lista delimitada (NÃO são permitidas duplicatas)); contar; count_distinct (ou seja, uma contagem dos valores únicos na coluna). Padrão: soma. Por exemplo: -c 5,4,6 -o sum,mean,count - dará a média da coluna 4, a soma da coluna 5 e a contagem da coluna 6. A ordem das colunas de saída corresponderá à ordem fornecida no comando.
Identifica se em dois conjuntos existe alguma da característica “sobreposta” ou não. Isso é conhecido como interseção de recursos e permite rastrear sobreposições entre dois conjuntos de características genômicas. Além disso, permite ter um controle preciso de como os cruzamentos são informados. Permite como entrada arquivos nos formatos BED, GFF, VCF e BAM.
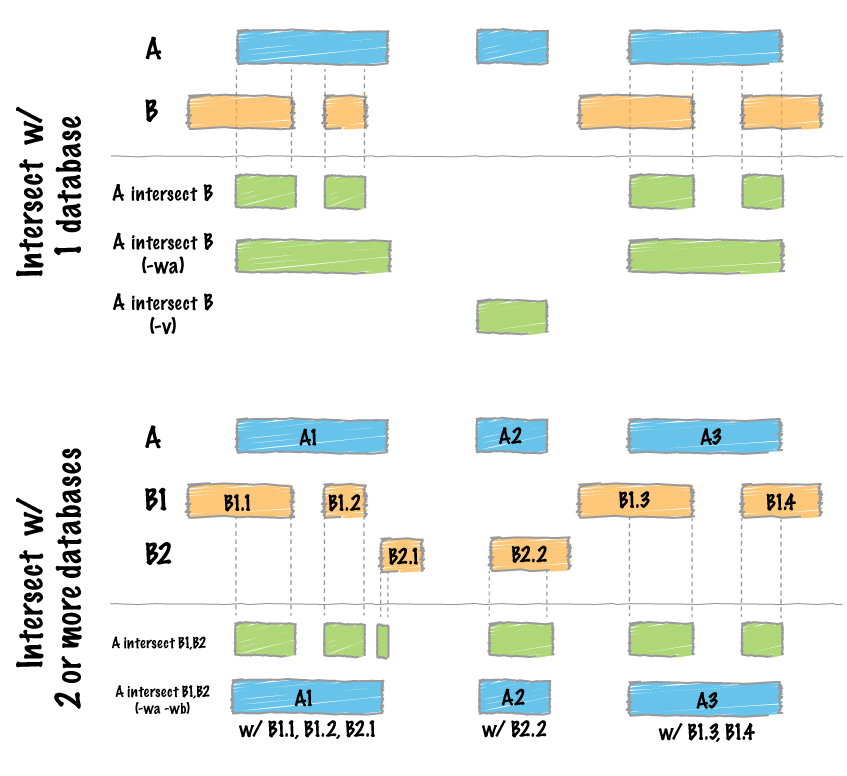
Exemplos:
Verificar anotações com 100% de sobreposição entre referência e query de forma reciproca:
bedtools intersect -a <fileA.gff> -b <fileB.gff> -s -f 1.0 -r > <output_file.gff>
-a: Entrada no formato BED/GFF/VCF/BAM.
-b: Entrada no formato BED/GFF/VCF/BAM.
-s: Considerar a fita, ou seja, relatar apenas acertos em B que se sobrepõem a A na mesma fita. Por padrão, as sobreposições são relatadas independentemente da vertente.
-f: Sobreposição mínima exigida como uma fração de A. O padrão é 1E-9 (ou seja, 1bp). -r: Exigir que a fração de sobreposição seja recíproca para A e B. Em outras palavras, se -f for 0,90 e -r for usado, isso requer que B se sobreponha a pelo menos 90% de A e que A também se sobreponha a pelo menos 90% de B.
Verificar anotações únicas da referência (em A):
bedtools intersect -a <fileA.gff> -b <fileB.gff> -v
-v: Relate apenas as entradas em A que não têm sobreposição em B. Como “grep -v”
Verificar anotações únicas da query (em B):
bedtools intersect -a <fileA.gff> -b <fileB.gff> -v
Para mais opções: bedtools intersect --help
A ferramenta calcula a profundidade e a amplitude da cobertura dos recursos do arquivo B nos recursos do arquivo A. Uma vantagem que oferece é que ele não apenas conta o número de recursos que se sobrepõem a um intervalo no arquivo A, mas também calcula a fração de bases no intervalo em A que foram sobrepostas por um ou mais recursos.
Chromosome ~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
BED FILE A *************** *************** ****** **************
BED File B ^^^^ ^^^^ ^^ ^^^^^^^^^ ^^^ ^^ ^^^^
^^^^^^^^ ^^^^^ ^^^^^ ^^
Result [ N=3, 10/15 ] [ N=1, 2/15 ] [N=1,6/6] [N=6, 12/14 ]
Vamos ao exemplo:
$ cat A.bed
chr1 0 100
chr1 100 200
chr2 0 100
$ cat B.bed
chr1 10 20
chr1 20 30
chr1 30 40
chr1 100 200
$ bedtools coverage -a A.bed -b B.bed
chr1 0 100 3 30 100 0.3000000
chr1 100 200 1 100 100 1.0000000
chr2 0 100 0 0 100 0.0000000
Linha de comando: bedtools coverage -a <fileA.bed> -b <fileB.bed > <output_file.gff>, onde:
-a: Entrada no formato BED/GFF/VCF/BAM.
-b: Entrada no formato BED/GFF/VCF/BAM.
Converta alinhamentos BAM para o formato BED
Linha de comando: bedtools bamtobed -i <file.bam> > <file.bed>, onde:
-i: Entrada no formato BAM.
Converte arquivos no formato BAM para o formato FASTQ
Linha de comando: bedtools bamtofastq -i <file.bam> -fq <file.fastq>, onde:
-i: Entrada no formato BAM.
-fq: Saída no formato FASTQ.
Converte recursos em um arquivo no formato BAM. Isso é útil como um meio eficiente de armazenar grandes anotações do genoma em um formato compacto e indexado para fins de visualização.
Linha de comando: bedtools bedToBam -i <file.bed> -g <genoma.fasta> > <output_file.bam>, onde:
-i: Entrada no formato BED/GFF/VCF.
-g: Genoma.fasta
Para mais opções do BEDTools: https://bedtools.readthedocs.io/en/latest/content/bedtools-suite.html
Quinlan, Aaron R., and Ira M. Hall. "BEDTools: a flexible suite of utilities for comparing genomic features." Bioinformatics 26.6 (2010): 841-842.
Agradeço a pesquisadora Mayla Abrahim Costa - FIOCRUZ (lattes: http://lattes.cnpq.br/4202137163257102) pela ajuda no desenvolvimento do conteúdo.