ref_cov3ma - ababaian/serratus GitHub Wiki
cov3ma
Reference pangenome associated with the summary/ and bam/ files on lovelywater2. Also referred to "nucleotide search" in the main Serratus manuscript.
- Sequence Files:
s3://lovelywater2/seq/cov3ma/ - Make notebook entry
Description
To create a collection of viral pangenomes, a comprehensive set of complete and partial genomes representing the genetic diversity of each vertebrate viral family, we used two approaches.
For Coronaviridae, we combined all RefSeq (n = 64) and GenBank (n = 37,451) records matching the NCBI Nucleotid server query "txid11118[Organism:exp]" (date accessed: June 1st 2020). Sequences <200 nt were excluded as well as sequences identified to contain non-CoV contaminants during preliminary testing (such as plasmid DNA or ribosomal RNA fragments). Remaining sequences were clustered at 99% identity with UCLUST and masked by Dustmasker with --window 30 and --window 64. The final query contained 10,101 CoV sequences.
For all other vertebrate viral family pangenomes, RefSeq sequences (n = 2,849) were downloaded from the NCBI Nucleotide server with the query "Viruses[Organism] AND srcdb_refseq[PROP] NOT wgs[PROP] NOT cellular organisms[ORGN] NOT AC_000001:AC_999999[PACC] AND ("vhost human"[Filter] AND "vhost vertebrates"[Filter])" (date accessed: May 17th 2020). Retroviruses (n = 80) were excluded as preliminary testing yielded excessive numbers of alignments to transcribed endogenous retroviruses. Each sequence was annotated with its taxonomic family according to its RefSeq record; those for which no family was assigned by RefSeq (n = 81) were designated as "unknown".
All AMR sequences associated with the CARD database were clustered at 95% nucleotide identity and included.
Old make notes:
Input and output
Input is 1. Cov0, Artem's catch-all set, and 2. the current set of Cov Refseq records.
Primary output is a FASTA file with the mapping reference, also called pan-genome (if one family) or mega-genome (if many families).
Meta-data files are also generated to document the procedure and assist in downstream analysis of alignments.
All Refseq sequences must be included in the output.
Every accession in Cov0 must be included or excluded for an explicit reason. As a check on the protocol it must generate a master.tsv file with at least two fields: 1. accession and 2. reason. Every accession must appear exactly once in master.tsv.
FASTA output includes complete genomes and fragments. Every fragment Q is annotated with the start position implied by an alignment to a full-length genome T. The start position is calculated as (start position in T of local alignment) - (number of unaligned bases at the start of Q).
Processing
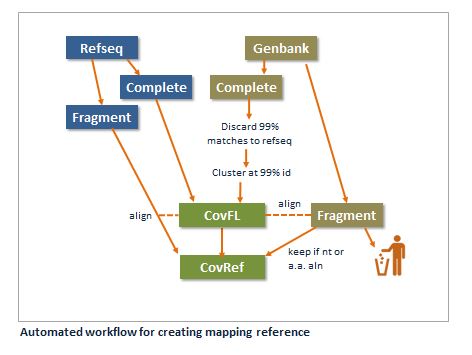
A sequence in Cov0 is either a complete genome or a fragment.
A sequence is a complete genome if the Genbank record has "complete genome" in the DEFINITION field. Anything else is a fragment, even if it is a complete something else, e.g. "complete cds".
Hierarchy of trust
Refseqs are most trusted. All refseqs are included in Covref. Second are full-length genomes. Fragments are included if they have a good nt or a.a. alignment to a complete genome. If no nt or a.a. alignment to a full-length sequence is found, a fragment is discarded.
Non-refseq complete genomes are removed if they have a global alignment with >=99% identity to a complete refseq genome. The remaining non-refseq complete genomes are clustered at 99% identity.
Refseq full-length genomes plus remaining non-refseq genomes after 99% clustering are combined to give a full-length set CovFL.
Fragments are aligned to CovFL. If a fragment has a local nt alignment with >=99% identity covering >=95% of the sequence, the fragment is discarded. Otherwise, if it has an alignment with >=70% identity with E-value <1e-6 it is kept. If it has neither, it is added to the Straggler set.
If a Straggler fragment has a tblastx alignment with no stop codons of length at least 64 a.a.'s to a CovFL sequence, it is kept. Otherwise, it is discarded.