4.5.2 Docking to allosteric binding sites... - Valdes-Tresanco-MS/AMDock-win GitHub Wiki
In this tutorial, we shall be working with a very interesting system, the M2 muscarinic acetylcholine receptor. Throughout the tutorial, we are going to recall some limitations related to scoring functions in docking programs as well as how AMDock can be used for studying allosteric binding in some receptors. Let's begin!
The M2 muscarinic receptor subtype plays a key role in modulating cardiac function and many important central processes, such as cognition and pain perception (1). As it was among the first GPCRs to be purified (2) and cloned (3), the M2 receptor has long served as a model system in GPCR biology and pharmacology (4). In this tutorial, we are going to use the active human M2 muscarinic acetylcholine receptor bound to the agonist iperoxo and allosteric modulator LY2119620 (PDB ID: 4MQT). This structure contains the orthosteric agonist iperoxo and the positive allosteric modulator LY2119620 simultaneously bound to the receptor. Let's see if we can predict correctly the binding modes of both ligands. Let's start with the agonist first!
Receptor (rec.pdb) and ligand (lig1.mol2) files are available at Installation_path/Doc/Tutorials/V_Additional_Tutorials/2.Docking_to_allosteric_binding_sites folder. Of note, the ligand coordinates have been randomized and the receptor contains the crystallographic ligand which coordinates will be used for defining the search space. In other words, we are going to perform a re-docking experiment. The crystal structure of the complex (complex.pdb) is available as well for comparison purposes.
- Open AMDock program
- Select Autodock Vina
- Set the Project Name (default Docking_Project)
- Set the Location for Project and push the button “Create Project”
- Check that “Simple Docking” checkbox is selected (It is checked by default)
- Choose the receptor file(Installation_path/Doc/Tutorials/V_Additional_Tutorials/2.Docking_to_allosteric_binding_sites/rec.pdb)
- Choose the ligand file (Installation_path/Doc/Tutorials/V_Additional_Tutorials/2.Docking_to_allosteric_binding_sites/lig1.mol2)
- Press the “Prepare Input” button
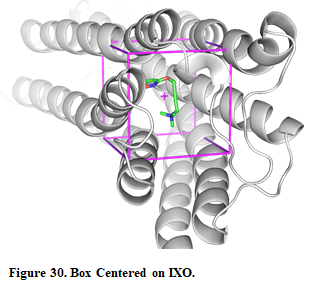
- Then, for defining a search space, pick “Center on Hetero” and select the ligand “A:IXO:501”
- Press “Define Search Space” button
Once the search space is defined, you can see the box in the receptor by pressing the button “Show in PyMOL”
Note: If you want to modify the box, you can use the AMDdock plugin in PyMOL. More details about this procedure here.
- Press the “Run” button
- When docking run ends, the “Results Analysis” tab will appear automatically. There, you will observe a summarizing table with Binding Energies, Estimated Ki values, and Ligand Efficiencies.
The number of poses and energies can vary since this is docking engine dependent.
- In the results table, just the first pose is selected. Press the button “Show in PyMOL” in the bottom left corner to visualize the complex.
If you want to see all the predicted poses, just select all the poses in the result table and press “Show in PyMOL”
- The crystal structure can be superposed on the predicted structure. Press “Open” in the upper left corner in PyMOL and find the X-ray structure (Installation_path/Doc/Tutorials/V_Additional_Tutorials/2.Docking_to_allosteric_binding_sites/complex.pdb)

As can be seen, Vina is able to predict correctly the agonist binding pose, although it tends to underestimate the binding energy (predicted~-6.5kcal/mol vs experimental~-11.4kcal/mol). The X-ray structure reveals a clear interaction between the polar head of the ligand and charged residues (D103). The ligand also interacts with aromatic residues (Y403 and 426) through the so-called pi-cation interaction. This kind of interaction is no well caught in scoring functions, though tending to underestimate the binding energy.
Let's try to predict the binding mode of the allosteric modulator!
Receptor (rec.pdb) and ligand (lig2.mol2) files are available at Installation_path/Doc/Tutorials/V_Additional_Tutorials/2.Docking_to_allosteric_binding_sites folder.
- Open AMDock program
- Select Autodock Vina
- Set the Project Name (default Docking_Project)
- Set the Location for Project and push the button “Create Project”
- Check that “Simple Docking” checkbox is selected (It is checked by default)
- Choose the receptor file(Installation_path/Doc/Tutorials/V_Additional_Tutorials/2.Docking_to_allosteric_binding_sites/rec.pdb)
- Choose the ligand file (Installation_path/Doc/Tutorials/V_Additional_Tutorials/2.Docking_to_allosteric_binding_sites/lig2.mol2)
- Press the “Prepare Input” button
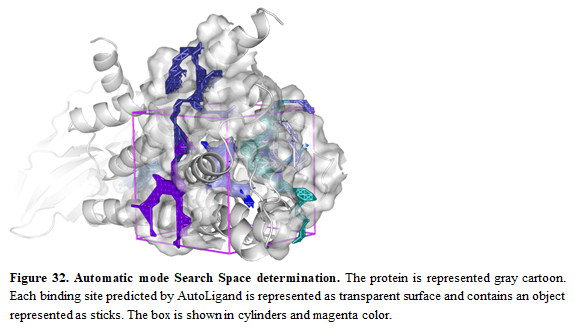
- Then, for defining a search space, pick “Automatic”
- Press “Define Search Space” button
Oftenly, allosteric binding sites are unknown and it's necessary to use some tools to predict possible allosteric sites. In this case, we aim to show that Autoligand can be used to do so. We also recommend checking other tools and double-checking the predicted sites from Autoligand. Once the search space is defined, you can see the box in the receptor by pressing the button “Show in PyMOL”. Multiple predicted binding sites from Autoligand will be shown
- Press the “Run” button
- When docking run ends, the “Results Analysis” tab will appear automatically. There, you will observe a summarizing table with Binding Energies, Estimated Ki values, and Ligand Efficiencies.
- In the results table, just the first pose is selected. Press the button “Show in PyMOL” in the bottom left corner to visualize the complex.
- The crystal structure can be superposed on the predicted structure. Press “Open” in the upper left corner in PyMOL and find the X-ray structure (Installation_path/Doc/Tutorials/V_Additional_Tutorials/2.Docking_to_allosteric_binding_sites/complex.pdb)
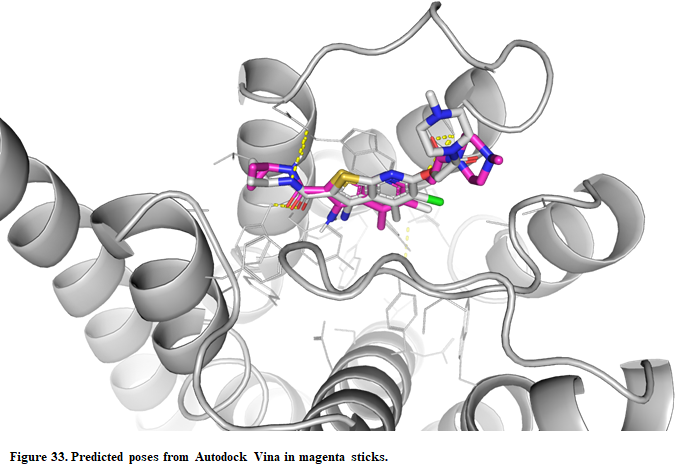
Vina was also able to predict the correct binding mode for the allosteric modulator. Although we succeeded in both cases, we need to be cautious when dealing with allosteric sites. Experimental validation is highly recommended through site-mutagenesis, X-ray, etc.