4.3 Off target docking - Valdes-Tresanco-MS/AMDock-win GitHub Wiki
For this tutorial, we selected as a study case the PI3K system, as in the previous cases. We intend to demonstrate how AMDock allows for a comparison between the two binding modes and predicted affinities for both complexes, i.e. target and off-target. The binding mode of the ligand SAR405 will be predicted for both the target (PDB ID:4UWH (Vps34)) and the off-target (PDB ID: 3APF (PI3Kγ)). It is known this inhibitor binds Vps34 preferentially with IC50=1.2nM, while IC50 for the other isoforms is > 10 000 nM.
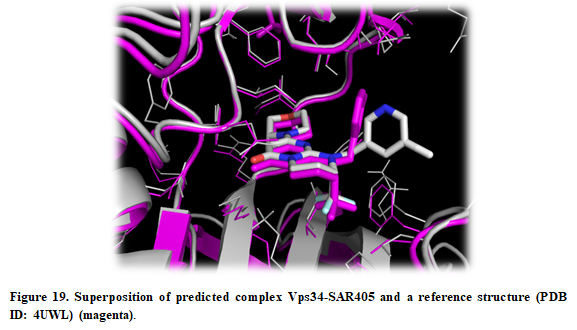
The receptors (4uwh.pdb and 3apf.pdb) and ligand (sar405.pdb) files are available at (Installation_path/Doc/Tutorials/II_Off-Target_Docking) folder. The structure of Vps34 complexed with SAR405 (complex.pdb, PDB ID:4OYS) is available as well for comparison purposes.
- Open AMDock program
- Select Autodock Vina
- Set the Project Name (default Docking_Project)
- Set the Location for Project and push the button "Create Project"
- Check that “Off-Target Docking” checkbox is selected
Automatically, a new text box for introducing off-target receptor will be available and as well as a new button “Align Proteins” which allows for aligning both receptors.
- Choose the target receptor file (Installation_path/Doc/Tutorials/II_Off-Target_Docking/4uwh.pdb)
- Choose the off-target receptor file (Installation_path/Doc/Tutorials/II_Off-Target_Docking/5ncy.pdb)
- Choose the ligand file (Installation_path/Doc/Tutorials/II_Off-Target_Docking/sar405.pdb)
- Press “Align Proteins”
This will be critical to define a similar search space in both proteins. The method used to align the proteins is quite simple and is performed by PyMOL. That way, it is mandatory to check the quality of the alignment. In general, both proteins should be similar, hence we should expect a low value of RMSD (you can check the log in the left side of the screen the RMSD value for the alignment).

- Press the “Prepare Input” button
- Then, for defining a search space, pick “Center on Hetero” and select the ligand “A:JXM:1876” for target receptor and “A:BMW:1103” for the off-target.
- Press “Define Search Space” button
Once the search space is defined, you can see the box by pressing the button “Show in PyMOL”. In this case, both receptors are aligned and represented as cartoon with ligands in sticks.

- Press “Run” button
- When docking run ends, the “Results Analysis” tab will appear automatically. There, you will observe a summarizing table with Binding Energies, Estimated Ki values, and Ligand Efficiencies. Also, the selectivity between the target and off-target receptors is shown.

- In each one of the tables, only the first pose is selected. Press the button “Show in PyMOL” in the bottom left corner to visualize the complex.
If you want to see all the predicted poses, just select all the poses in the result table and press “Show in PyMOL”. On the other hand, if you want only to visualize one of the complexes, use Ctrl+click to deselect the poses you are not interested in.
- The crystal structure can be superposed on the predicted structure. Press “Open” in the upper left corner in PyMOL and find the X-ray structure (Installation_path/Doc/Tutorials/II_Off-Target_Docking/complex.pdb).
- A detailed report of the entire process can be seen on the right side of the screen, which allows for tracking all the results from different programs and algorithms. This log file can be saved in case an error occurred and sent to the developers.