For contributors - xinwang2hms/SPP GitHub Wiki
First of all, welcome to get on board and thank you for your time and effort to contribute to the package! To get a quick start, please go through the demo of ENCODE data analysis. To further facilitate your process of contributing, here are some information about the design of the new spp package. Don't hesitate to contact me if you find any bug or if you have any suggestion. It is always better to make changes before we release and before people start to use.
To do list
Here are some changes we plan to finish in the next update:
-
Addressing two warnings about c++ code in
R CMD check. -
Finishing all documentation: wiki, release website, R package help files and vignette.
-
New class
ChIPSeq. This class is aimed for representing a batch of ChIP-seq experiments including multiple replicates under multiple conditions for multiple IPs. -
Quality control. Two kinds of QC are now planned to be added: (A) QC to detect phantom peaks and quantify the quality of each individual experiment. This functionality should be added as a new method to the class of
AlignedTags. (B) QC for assessing the consistency between replicates. This functionality should be added as a method to the new classChIPSeq. -
Additional ChIP-seq profiles such as metagene profiles.
Naming convention
In the new spp package, different naming conventions are adopted to distinguish classes, methods/functions and arguments:
-
Class names are composed of words joined without spaces, with each word's initial letter in capitals (e.g. AlignedTags, ChIPSeqProfile).
-
Method/function names are composed of words connected with dots (e.g. set.param, write.tdf).
-
Argument names are composed of words joined with underscores (e.g. window_size, sort_by).
Object-oriented programming
The core of spp consists of reference classes, which are relatively new to R. The main advantage over other OO systems in R lies in its ability to pass objects by reference instead of value, which can greatly reduce memory usage and improve computational efficiency. The programming style of reference classes are very different from S4 and S3 classes. So getting familiar with this new OOP system before you program is highly suggested.
Introduction of classes
The class diagram (not pipeline) below illustrates all reference classes and their relationships.
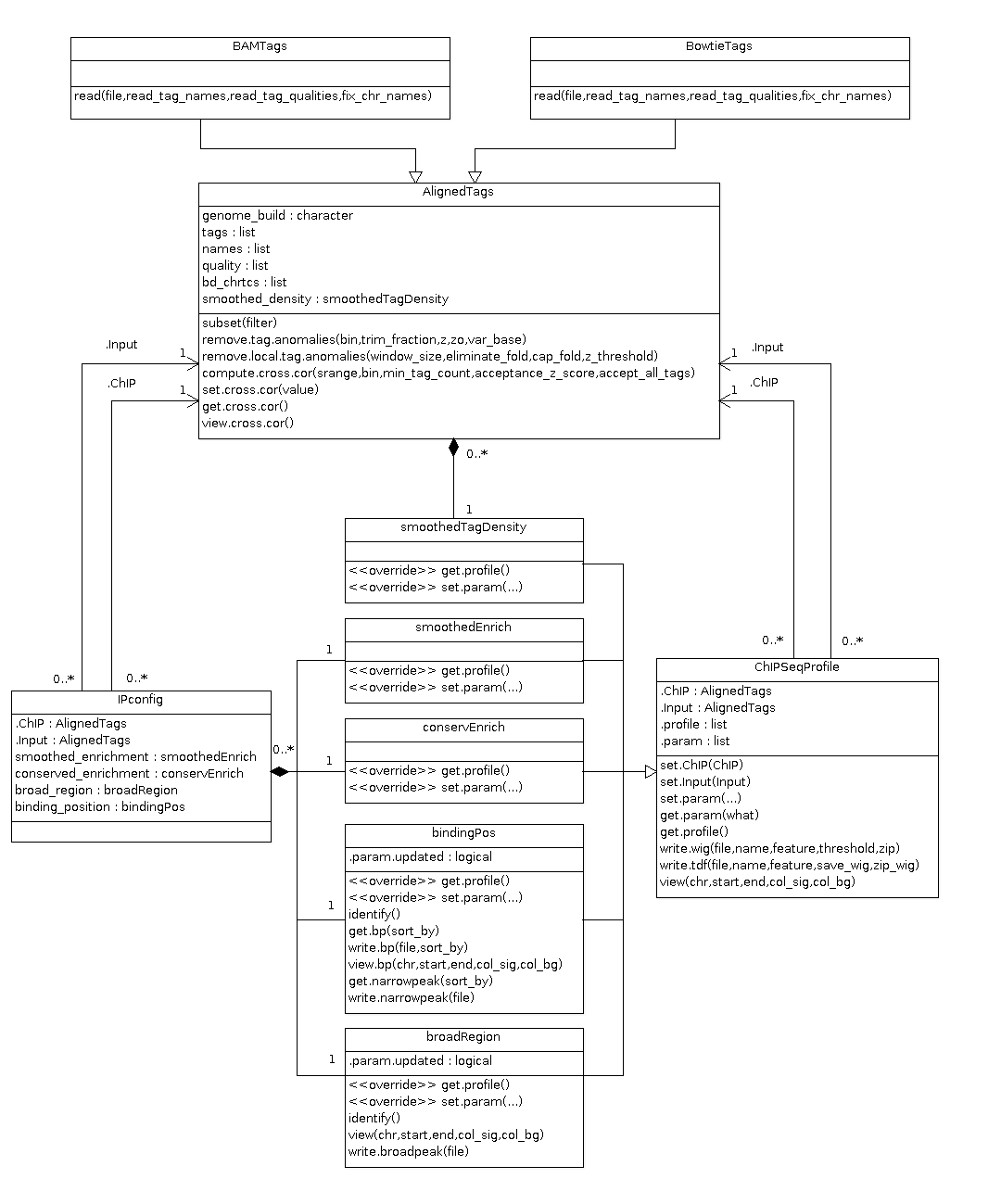
Basically, there are 3 types of classes:
-
Classes representing aligned tags.
AlignedTagsis the root class which includes common attributes such asgenome_build,tags,bd_chrtcs(binding characteristics) andsmoothed_density(smoothed tag density profile). It also includes common functionalities such ascompute.cross.corandremove.tag.anomalies. However, this class should not be directly called, as there is no method to import ChIP-Seq data. Instead, classBAMTagsandBowtieTags, which are specializations of classAlignedTags, should be used to create objects to handle ChIP-seq data. Currently, spp only supports BAM and Bowtie alignment results. If you want to add more functionalities to these classes, please go to those R files and add new methods as you want. If you need to import aligned tags resulted from a different alignment tool, please write a new class, which should also depend on theAlignedTagsclass. If you think that the structure of these classes should be modified (e.g. add a new attribute or restructure the class), please let me know and we have a discussion. -
Classes describing ChIP-seq profiles. These classes also include a root class
ChIPSeqProfileand its various specializations. These subclasses aresmoothedTagDensity,smoothedEnrich,conservEnrich,bindingPosandbroadRegion.ChIPSeqProfilegeneralizes common properties of different profiles and has basic versatile methods to set parameters, write profiles to wig/tdf files, preview profiles in a specific genomic region, etc. More specific functionalities are implemented in subclasses. For instance, in classbindingPos, there are methods to get binding positions and narrow peaks. If you want to add a new type of ChIP-seq profile, please go ahead to add a new class, which should be dependent on classChIPSeqProfile. If you want to add new functionalities to existing classes, just add new methods within corresponding R files. Again, please notify me if you want to modify the basic design of classes (e.g. class attributes), as those changes can affect a lot of downstream classes and methods. -
The
IPconfigclass. This class represents a typical IP configuration, one ChIP one Input, in ChIP-seq experiments. It contains references to two objects of classAlignedTags, representing a ChIP and a Input data set. Therefore, the class itself does not copy but refer to existingAlignedTagsobjects, which should save lots of memory. For the same reason, however, please be aware of modification on thoseAlignedTagsobjects, as it affectsIPconfigobjects. The functionalities ofIPconfigare actually realized through objects of various ChIP-seq profiles included as attributes in the class, so changes should mainly be made to those specific ChIP-seq profile classes. If you do think that the classIPconfigneeds some changes, please notify me.
###Updating saved objects
Saved reference objects will not update methods automatically every time you revise methods of classes. The existing classes in spp are designed in a way that the initialization methods can recognize both 'copy' object and 'new' object. For instance, in the demo we can create a new object of AlignedTags by the following code:
> encH3K4 <- BowtieTags(genome_build="mm9")
> encH3K4$read(file="H3K4me3.btm.bz2")
The code above 'newed' an object. However, we can also create an object by copying an existing object, e.g.:
> encH3K4 <- BowtieTags(encH3K4)
Note that in the new encH3K4, methods are up-to-date already.