Pipeline management: SnakeMake - ua-datalab/Bioinformatics GitHub Wiki
Image credits: wikimedia.
{kind=link}
Important
🕐 Schedule
- 3:00pm-3:15pm: Welcome and introdution to topic (What is Snakemake?)
- 3:15pm-3:50pm: Understanding Snakemake Syntax and pipeline execution
- 3:50pm-4:00pm: Resources and closing remarks
Important
❗ Requirements
- Basic command line knowledge
- Access to a Terminal
- Unix and Mac users already have access to the Terminal
- Windows users can use either PowerShell or the Windows Subsystem for Linux (WSL)
- A registered CyVerse account (Register for a CyVerse account)
Important
✅ Expected Outcomes
- Understand the concept and significance of workflow management in bioinformatics.
- Identify the advantages of using Snakemake for pipeline management.
- Break down and navigate the Snakemake syntax and structure.
- Break down a practical Nextflow script and explain its components.
- Execute a basic Snakemake pipeline.
Welcome to the Snakemake workshop! In this session, we'll explore how Snakemake streamlines the process of creating reproducible and scalable bioinformatics workflows. You'll learn about essential concepts such as rules, dependencies, and directed acyclic graphs (DAGs) that structure your analyses.
Important
This workshop uses is an adaptation of the official Snakemake basics tutorial. This workshop is created to cover the basics, there is a lot more to learn on Snakemake.
Note
Overview of Pipeline As this can be classified as an analysis pipeline, it is important for us to highlight what the major steps are:
[Mapping to reference (bwa)]
↓
[Sorting alignments (samtools)]
↓
[Indexing alignments (samtools)]
↓
[Calling variants (bcf)]
↓
[generate quality score plot (python)]

Image credits: Math Lady meme.
In order to get things started:
- Execute the JupyterLab Bioscience CyVerse App and open the Terminal with 4 CPU cores (min and max) and 4GB RAM memory.
- Today's workshop uses Snakemake's official tutorial and data. For simplicity, a couple of files have been added to ensure quick execution (
scripts/plot-quals.py,snakefile). Clone the git repository to the App (does not require a GitHub account):and move into the cloned repositorygit clone https://github.com/CosiMichele/snakemake-tutorial-data.gitcd snakemake-tutorial-data
In case you cannot use CyVerse, you can run this code on GitPod, directly from the Repository.
You are now ready for the workshop!

Image source: Felix Mölder et al., Sustainable data analysis with Snakemake, F1000 Research, 2021.
Snakemake is a powerful workflow management system designed for creating reproducible and scalable data analyses. It allows users to define workflows in a concise and human-readable format using a simple domain-specific language. Here are some key features and aspects of Snakemake:
- Declarative Syntax: Users define rules that describe how to produce output files from input files, making it easy to specify dependencies and the order of execution.
- Directed Acyclic Graph (DAG): Snakemake automatically constructs a DAG from the defined rules, ensuring tasks are executed in the correct order based on their dependencies.
- Reproducibility: By using a consistent environment for each rule, often defined with Conda or Docker, Snakemake helps ensure that analyses can be reliably reproduced.
- Parallel Execution: Snakemake can run multiple tasks in parallel, utilizing available resources effectively to speed up the workflow execution.
- Resource Management: Users can specify resource requirements (like CPU and memory) for each task, allowing for better utilization of computational resources.
- Scalability: Snakemake is suitable for workflows ranging from small scripts to large-scale analyses, and it can be executed on various platforms, including local machines, clusters, and cloud environments.
Snakemake was launched in the early 2020s and has seen rapid growth and use in the bioinformatic fields. Snakemake adds a layer of readability due to its closeness to the Python programming language, as Snakemake is built on Python and utilizes its syntax and functionalities.
Due to its closeness to Python, Snakemake is often cited as a great integrator of Conda, an extremely popular package and enviroment manager hosting python and other non-python software (e.g., BLAST, BWA, Samtools, etc).
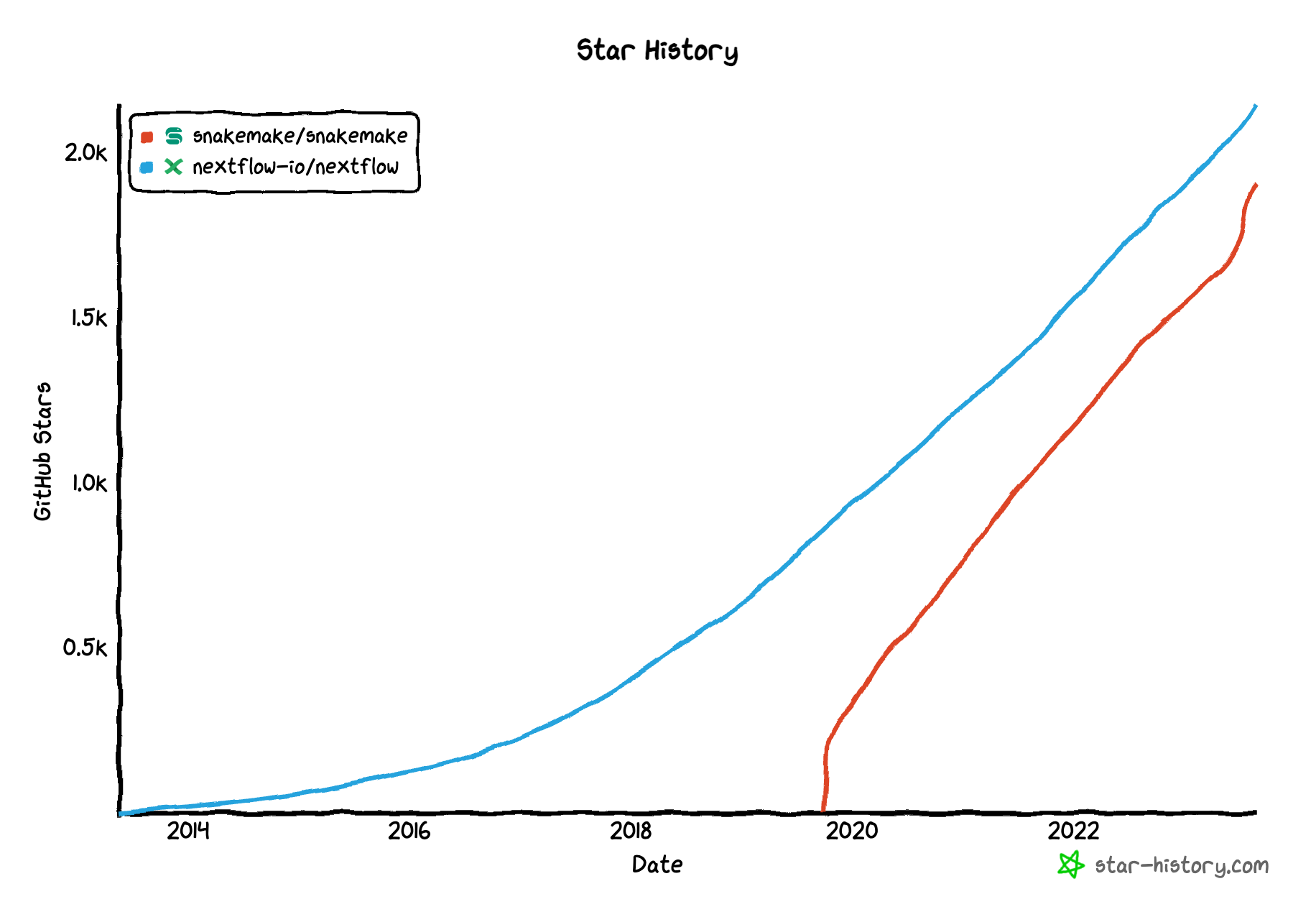
Image source: Noah Lebovic, Nextflow vs Snakemake, flowdeploy CEO; the image depics the repositories' GitHub star count (aka "followers", but for geeks and nerds).

Image source: Felix Mölder et al., Sustainable data analysis with Snakemake, F1000 Research, 2021.
Snakemake's main features revolve around Rules, with each rule having an input, output, (facultatively) a conda directive, and a shell OR a script. Additionally, Snakemake has a special rule: the All rule. This ensures that the workflow is done by checking on the last expected output. Wildcards, DAG (Directed Acyclic Graph) are two other important components to any Snakemake workflow.
Here is the example pipeline from the snakemake official documentation:
Click here for the raw NextFlow code
SAMPLES = ["A", "B"]
rule all:
input:
"plots/quals.svg"
rule bwa_map:
input:
"data/genome.fa",
"data/samples/{sample}.fastq"
output:
"mapped_reads/{sample}.bam"
shell:
"bwa mem {input} | samtools view -Sb - > {output}"
rule samtools_sort:
input:
"mapped_reads/{sample}.bam"
output:
"sorted_reads/{sample}.bam"
shell:
"samtools sort -T sorted_reads/{wildcards.sample} "
"-O bam {input} > {output}"
rule samtools_index:
input:
"sorted_reads/{sample}.bam"
output:
"sorted_reads/{sample}.bam.bai"
shell:
"samtools index {input}"
rule bcftools_call:
input:
fa="data/genome.fa",
bam=expand("sorted_reads/{sample}.bam", sample=SAMPLES),
bai=expand("sorted_reads/{sample}.bam.bai", sample=SAMPLES)
output:
"calls/all.vcf"
shell:
"bcftools mpileup -f {input.fa} {input.bam} | "
"bcftools call -mv - > {output}"
rule plot_quals:
input:
"calls/all.vcf"
output:
"plots/quals.svg"
script:
"scripts/plot-quals.py"
Note
Construction Worksite Analogy
Imagine Snakemake as a construction manager overseeing the construction of a large building. Each part of the building represents a rule, and the materials, tools, and workers represent the inputs, outputs, and shell commands needed to complete the project.

Rules are Construction Tasks

- Each rule in Snakemake is like a specific construction task. It defines the materials (inputs) needed to complete the task, the finished part of the building (output), and the work instructions (shell command) to get the job done.
- Example: Pouring the foundation requires cement, water, and gravel, which results in a finished concrete base.
DAG is the Construction Blueprint

- The DAG (Directed Acyclic Graph) is like the blueprint that the construction manager follows. It maps out the entire project, showing how each task (rule) fits into the larger plan, ensuring nothing is missed and that tasks are completed in the right order.
- Example: The blueprint shows that electrical wiring should be installed after the walls are built but before the drywall goes up.
The "All" Rule is the Completed Building

Just as the manager ensures that every part of the building is finished before calling it complete, the "all" rule in Snakemake guarantees the entire workflow is executed and all final outputs are produced. Example: the completed building!
There are other processes that are required in Snakemake that are missing from this larger picture:
- Parallelization: multiple teams working on different aspect of a task (one team pours concrete, the other lays it).
- Resources Allocation (& Conda Environments): specialized tools and workers that are accessed when and where needed (an electrician is potentially not required when first levelling ground).
- Dependencies are Construction Phases: Just like you can’t install the roof before building the walls, some tasks depend on others being completed first. Snakemake ensures that each task happens in the right order.
Rules are the building blocks of a Snakemake workflow. Each rule defines how to create an output file from input files using a shell command.
Example:
rule bwa_map:
input:
"data/genome.fa",
"data/samples/{sample}.fastq"
output:
"mapped_reads/{sample}.bam"
shell:
"bwa mem {input} | samtools view -Sb - > {output}"
The all rule specifies the final outputs of the entire workflow. This is the "target" rule that tells Snakemake what the end products of the workflow should be. It can be placed in the beginning or the end of the workflow (in the early iterations of Snakemake this was something that was strictly needed at the beginning of the workflow).
Example:
rule all:
input:
"plots/quals.svg"
Notice how in the example above, the expected output is defined as input. This is because of how Snakemake works, my having rule all "check" on the final output by ingesting it (thus requiring to be defined as input).
Snakemake automatically constructs a Directed Acyclic Graph (DAG) from the defined rules. The DAG represents the dependencies between rules, showing which rules need to be run in sequence and which can run in parallel. The DAG ensures that the workflow executes only the necessary steps, allowing Snakemake to avoid re-executing tasks if their outputs are up-to-date.
DAG can also be generated in an image format (svg) to help you visualize your pipeline. You can create the DAG image with snakemake --dag | dot -Tsvg > dag.svg.
Here is the representation of the example code's DAG:

Wildcards make Snakemake workflows dynamic by allowing file names to vary depending on a pattern. This helps automate workflows for multiple samples without repeating code. These are found throughout the code using brackets (e.g., {sample}).
These are established at the beginning of the script (e.g., SAMPLES = ["A", "B"], line 1 in our example script).
Popular Snakemake commands include:
- Basic workflow execution: this command executes the workflow defined in the Snakefile, building the output files specified in the rule all.
You can specify a target file with
snakemakesnakemake <target>. - Dry run: extremely useful to test the consistency of the pipeline without creating outputs; time saver.
snakemake -n - Specify the cores to be used:
or
snakemake --cores <number of cores>snakemake -j <number of cores> - Specify a configuration file (we have not covered
config.yaml, explanation here and here)snakemake --configfile <config.yaml> - Creating a pipeline diagram (DAG):
snakemake --dag | dot -Tpng > dag.png
In this workshop, we are going to execute a dry run, create a DAG diagram, and running the pipeline. The commands to execute in order are:
snakemake -nsnakemake --dag | dot -Tpng > dag.pngsnakemake --cores 4
1. Sample List Declaration
SAMPLES = ["A", "B"]
This line defines a list called SAMPLES, containing the sample identifiers "A" and "B". This variable is later used to expand rules for each sample, allowing for scalable processing of multiple samples without hardcoding paths.
2. Final Target Rule
rule all:
input:
"plots/quals.svg"
The rule all specifies the final output file that the entire workflow aims to produce: plots/quals.svg. This serves as a target for Snakemake, indicating what the workflow should ultimately generate. Snakemake will automatically determine all required intermediate steps to produce this file.
3. Mapping Reads with BWA
rule bwa_map:
input:
"data/genome.fa",
"data/samples/{sample}.fastq"
output:
"mapped_reads/{sample}.bam"
shell:
"bwa mem {input} | samtools view -Sb - > {output}"
The bwa_map rule performs read mapping using the BWA (Burrows-Wheeler Aligner) tool, using bwa mem to align the reads and pipes the output to samtools view to convert it into BAM format.
- Input: It takes the reference genome (
data/genome.fa) and the fastq file for a specific sample (denoted by{sample}). - Output: The output is a BAM file (
mapped_reads/{sample}.bam), which contains the mapped reads.
4. Sorting Mapped Reads
rule samtools_sort:
input:
"mapped_reads/{sample}.bam"
output:
"sorted_reads/{sample}.bam"
shell:
"samtools sort -T sorted_reads/{wildcards.sample} "
"-O bam {input} > {output}"
The samtools_sort rule sorts the mapped BAM files using samtools sort, with the -T option specifying a temporary file location based on the sample name.
- Input: It takes the BAM file produced by the bwa_map rule.
- Output: The output is a sorted BAM file (
sorted_reads/{sample}.bam). Execution: The command
5. Indexing Sorted BAM Files
rule samtools_index:
input:
"sorted_reads/{sample}.bam"
output:
"sorted_reads/{sample}.bam.bai"
shell:
"samtools index {input}"
The samtools_index rule creates an index for the sorted BAM files, simply calling samtools index to generate the index.
- Input: It takes the sorted BAM file from the samtools_sort rule.
- Output: The output is the index file (
sorted_reads/{sample}.bam.bai), which allows for efficient access to the BAM file during analysis.
6. Calling Variants with BCFtools
rule bcftools_call:
input:
fa="data/genome.fa",
bam=expand("sorted_reads/{sample}.bam", sample=SAMPLES),
bai=expand("sorted_reads/{sample}.bam.bai", sample=SAMPLES)
output:
"calls/all.vcf"
shell:
"bcftools mpileup -f {input.fa} {input.bam} | "
"bcftools call -mv - > {output}"
The bcftools_call rule performs variant calling on the sorted BAM files, using bcftools mpileup to generate the necessary data for variant calling and pipes it to bcftools call to produce the VCF.
- Input: It requires the reference genome, as well as the sorted BAM and index files for all samples (using expand to generate the paths for both samples "A" and "B").
- Output: The output is a VCF file (
calls/all.vcf), which contains the variant calls. Execution:
7. Plotting Quality Scores
rule plot_quals:
input:
"calls/all.vcf"
output:
"plots/quals.svg"
script:
"scripts/plot-quals.py"
The plot_quals rule generates a plot of quality scores from the variant calls. Instead of a shell command, this rule runs a Python script (scripts/plot-quals.py) that generates the plot based on the input VCF data.
- Input: It takes the VCF file generated by the bcftools_call rule.
- Output: The output is a plot in SVG format ("plots/quals.svg").

Snakemake is a powerful tool that will allow you to keep your pipelines clean and reproducible. Its extra layer of simplicity in comparison to Nextflow makes it less daunting and more easiliy picked up. It however isn't as versed in its parallelization and job scheduling as Nextflow is (also mentioned in their official publication) -- however this issue may already have been addressed.
Regardless of (potentially) minor issues, Snakemake is a great tool to learn for every bioinformatician needing to implement a workflow manager.
Resources: