Pipeline management: NextFlow - ua-datalab/Bioinformatics GitHub Wiki

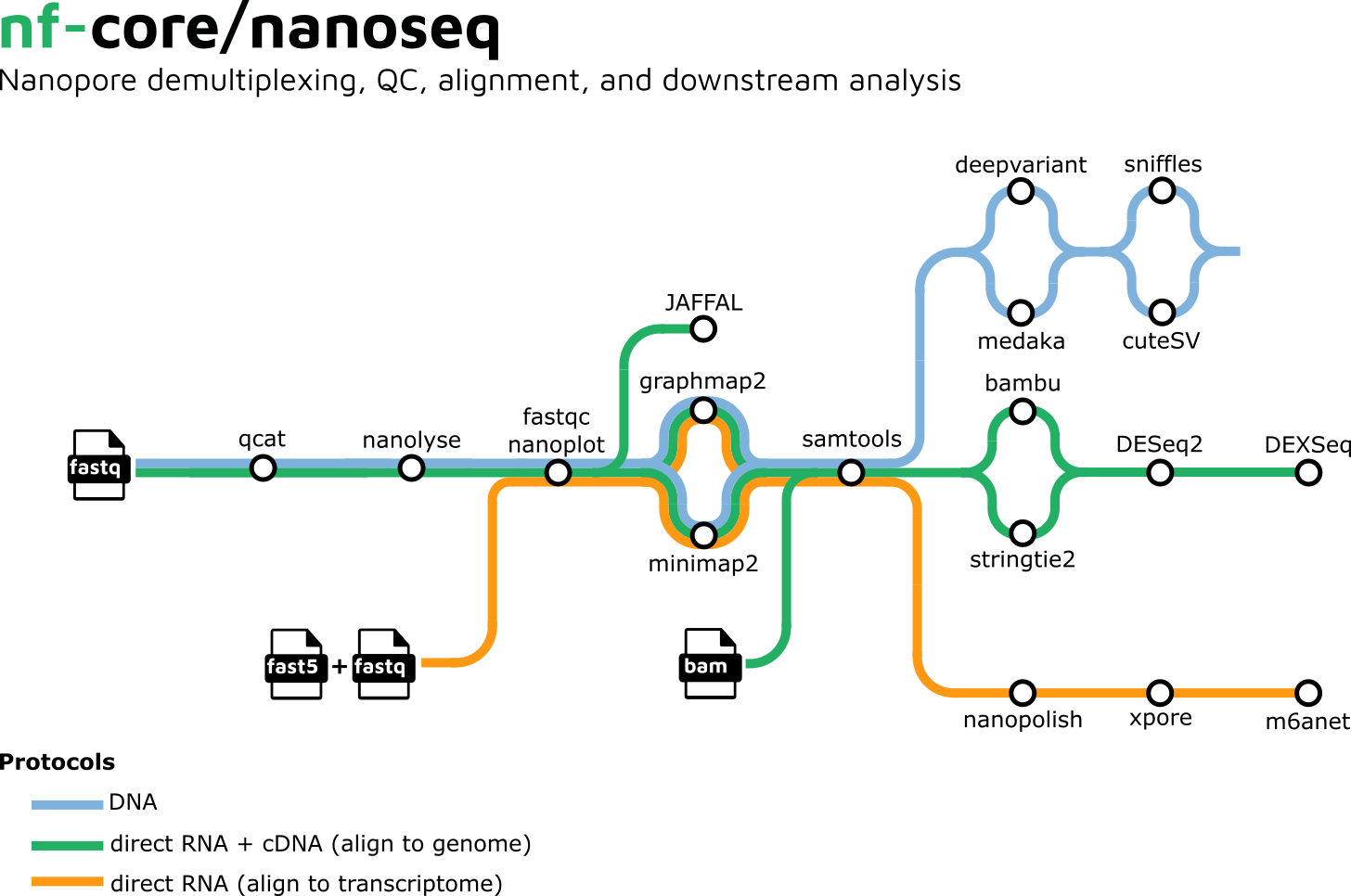
Image credits: nf-core/nanoseq
Important
🕐 Schedule
- 3:00pm-3:10pm: Welcome and introdution to topic (What is Nextflow?)
- 3:10pm-3:20pm: The importance of pipeline management and the direction of bioinformatics
- 3:20pm-3:50pm: Understanding Nextflow Syntax and pipeline execution
- 3:50pm-4:00pm: Resources and closing remarks
Important
❗ Requirements
- Basic command line knowledge
- Access to a Terminal
- Unix and Mac users already have access to the Terminal
- Windows users can use either PowerShell or the Windows Subsystem for Linux (WSL)
- A registered CyVerse account (Register for a CyVerse account)
Important
✅ Expected Outcomes
- Understand the concept and significance of workflow management in bioinformatics.
- Identify the advantages of using Nextflow for data-driven analysis.
- Navigate the Nextflow syntax and structure, including processes, channels, and workflows.
- Break down a practical Nextflow script and explain its components.
- Execute a basic RNA-Seq analysis pipeline, integrating tools like FastQC, Salmon, and MultiQC.
This workshop is designed to introduce participants to Nextflow, a powerful workflow management system that enables the creation, execution, and sharing of reproducible computational pipelines in bioinformatics. Participants will learn the fundamentals of Nextflow, including its importance in modern research, its unique features, and how it compares to other workflow management tools.
Important
This workshop uses is an adaptation of the Sequera Nextflow tutorial, shortened in order to fit time limits. Nextflow is something that cannot be learned in 1 hour or less, but its components are worth understanding . For this tutorial, the organism used is G. gallus, the red junglefoul (the o.g. chicken 🐔). All files (transcriptome, fastq files) are made available on CyVerse.
Note
Overview of Pipeline As this can be classified as an analysis pipeline, it is important for us to highlight what the major steps are:
[Indexing trascriptome]
↓
[Perform QC]
↓
[Perform quantification]
↓
[Create MultiQC report]

Image credits: It's Always Sunny in Philadelphia.
In order to get things started:
-
Execute the JupyterLab Bioscience CyVerse App and open the Terminal with 16 CPU cores (min and max) and 16GB RAM memory.
-
Initiate GoCommands, the CyVerse in-house transfer tool using
gocmd init.- Press enter (don't input anything) when propted for iRODS Host, Port, Zone; put your CyVerse username when asked for iRODS Username and your CyVerse password when asked for iRODS Password (Note: you will not see the password being typed as per standard Ubuntu security).
-
Copy the genome files using the following command:
gocmd get --progress /iplant/home/shared/cyverse_training/datalab/biosciences/nextflow_tutorial/This will copy:
- The G. gallus transcriptome (
transcriptome.fa) - Gut pair-end reads (
gut_1.fq,gut_2.fq) - The NextFlow code required for the workshop.
- Other read files for exercise purposes (liver, lung)
- The G. gallus transcriptome (
You are now ready for the workshop!
Click here for the raw NextFlow code
nextflow.enable.dsl=2
/*
* pipeline input parameters
*/
params.reads = "$baseDir/data/ggal/gut_{1,2}.fq"
params.transcriptome = "$baseDir/data/ggal/transcriptome.fa"
params.multiqc = "$baseDir/multiqc"
params.outdir = "results"
println """\
R N A S E Q - N F P I P E L I N E
===================================
transcriptome: ${params.transcriptome}
reads : ${params.reads}
outdir : ${params.outdir}
"""
.stripIndent()
/*
* create a transcriptome file object given the transcriptome string parameter
*/
transcriptome_file = file(params.transcriptome)
/*
* Define the `index` process that creates a binary index
* from the transcriptome file
*/
process index {
conda 'bioconda::salmon'
input:
path transcriptome_file
output:
path 'index'
script:
"""
salmon index --threads ${task.cpus} -t ${transcriptome_file} -i index
"""
}
/*
* Define a channel for paired reads and set the channel name to `read_pairs_ch`
*/
Channel.fromFilePairs(params.reads)
.ifEmpty { error "Cannot find any reads matching: ${params.reads}" }
.set { read_pairs_ch }
/*
* Define the `quantification` process that runs Salmon quantification
*/
process quantification {
conda 'bioconda::salmon'
input:
path index
tuple val(pair_id), path(reads)
output:
path "${pair_id}_quant"
script:
"""
salmon quant --threads ${task.cpus} --libType=U -i ${index} -1 ${reads[0]} -2 ${reads[1]} -o ${pair_id}_quant
"""
}
/*
* Define the `fastqc` process that performs quality control on reads
*/
process fastqc {
conda 'bioconda::fastqc'
tag "FASTQC on ${sample_id}"
input:
tuple val(sample_id), path(reads)
output:
path "fastqc_${sample_id}_logs"
script:
"""
mkdir fastqc_${sample_id}_logs
fastqc -o fastqc_${sample_id}_logs -f fastq -q ${reads}
"""
}
/*
* Define the `multiqc` process that generates a report from fastqc and quant outputs
*/
process multiqc {
publishDir params.outdir, mode: 'copy'
conda "bioconda::multiqc"
input:
path fastqc_files
path quant_files
output:
path 'multiqc_report.html'
script:
"""
multiqc .
"""
}
/*
* Workflow definition that orchestrates the execution of processes
*/
workflow {
/*
* Pass the transcriptome file to the `index` process
*/
index_ch = index(transcriptome_file)
/*
* Run the processes in the desired order and collect outputs
*/
fastqc_ch = fastqc(read_pairs_ch)
quant_ch = quantification(index_ch, read_pairs_ch)
multiqc(fastqc_ch, quant_ch)
}

Image source: nf-core/rnaseq.
Written in Groovy, Nextflow is an open-source workflow management system that allows researchers to write and execute data-driven computational workflows. It is designed to handle complex workflows in a scalable and reproducible manner.
Although Nextflow is heavily used in the bioinformatics field, its processes are used in other fields including cloud computing, data science, image analysis and machine learining.
- Portability: Nextflow can run on various platforms, including local machines, clusters, and cloud environments, without modification to the workflow script.
- Scalability/Parallel Execution: It automatically parallelizes tasks, optimizing resource utilization and speeding up the analysis.
- Reproducibility: Nextflow enables the creation of workflows that can be easily shared and rerun, ensuring that analyses can be replicated by others.
Uniquely to Nextflow is its ability to manage piplelines using Channels: these are processes that "activate" and remain active until the workflow is turned off. Other pipeline managers fail at this as these are reliant on outputs and inputs. Example: when you turn the water on and off, the pipes don't disappear.
Additionally, Nextflow has a robust community that create, share and maintain their pipelines at nf-co.re with extremely well documented work, responsive support and in-person and online events.
Nextflow has seen a surge in industry use, with Nanopore officially creating Nextflow pipelines through "EPI2ME Workflows". A great example of one of these pipelines is nanoseq: an analysis pipeline for Nanopore DNA/RNA sequencing data that can be used to perform basecalling, demultiplexing, QC, alignment, and downstream analysis.
Snakemake🐍: similar to Nextflow as both are flexible, scalable and portable workflow managers. Snakemake is preferred in many ways as it is similar to the Python coding language, offering a lesser steep learing curve (python vs groovy). Snakemake will be covered in more details later in the workshop series.
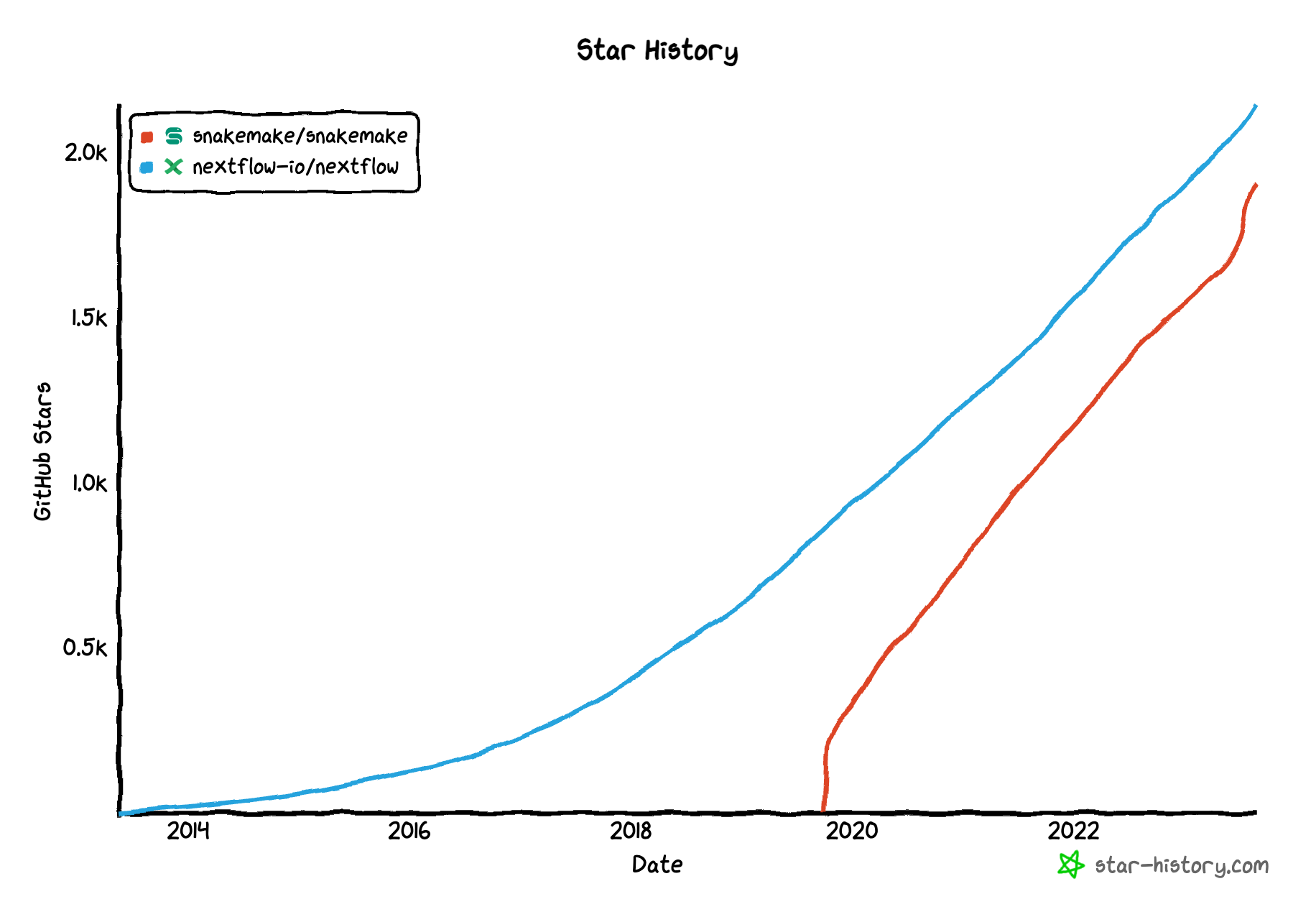
Image source: Noah Lebovic, Nextflow vs Snakemake, flowdeploy CEO; the image depics the repositories' GitHub star count (aka "followers", but for geeks and nerds).
Galaxy ✨: If you have been around bioinformatics for a while, it is likely that you have run into Galaxy. Its web platform allows for people to create and manage workflows with simplicity, and it is still to the day one extremely popular in the field. It is however limited in scalability, rendering larger and more complex workflows challenging.
There really isn't a single answer to all problems. Knowing what resources are available for your research needs can put you many steps ahead of the curve.
Nextflow has a number of complex components that need to be understood prior to using or writing a pipeline: Processes, Channels, Workflows, Directives.
A process defines a task that performs computations or manipulations on data. Each process specifies inputs, outputs, and a script to run.
process processName {
input:
// Define inputs
output:
// Define outputs
script:
// The command or script to execute
}
Example:
process align {
input:
path reads
output:
path "aligned_${reads.getName()}"
script:
"""
bowtie2 -x reference_genome -U ${reads} -S aligned_${reads.getName()}
"""
}
Channels are used to communicate data between processes. They can be used to collect data from files, create lists, or combine outputs from multiple processes.
Channel.fromFilePairs('path/to/files')
Example:
Channel
.fromFilePairs('data/sample_{1,2}.fastq')
.ifEmpty { error "No reads found!" }
.set { read_pairs_ch }
A workflow orchestrates the execution of processes and defines the data flow between them. It combines processes and channels to form a complete analysis pipeline.
workflow {
// Call processes and connect channels
processA(input_channelA)
processB(input_channelB)
}
Example:
workflow {
// Define the workflow
read_pairs_ch = read_pairs_channel()
align(read_pairs_ch)
}
Directives are special annotations used to control the behavior of processes and workflows. They can specify resource requirements, execution environments, or specific behavior.
process processName {
directive 'value'
}
Example:
process align {
conda 'bioconda::bowtie2'
publishDir 'results/aligned'
input:
path reads
output:
path "aligned_${reads.getName()}"
script:
"""
bowtie2 -x reference_genome -U ${reads} -S aligned_${reads.getName()}
"""
}
The usual order of the components in a Nextflow script is:
- Parameters
-
Processes
- As many processes as needed; each process has its own Directives.
- Workflow
Altogether these come to make the following mock-pipeline:
nextflow.enable.dsl=2
params.reads = 'data/sample_{1,2}.fastq'
process align {
conda 'bioconda::bowtie2'
publishDir 'results/aligned'
input:
path reads
output:
path "aligned_${reads.getName()}"
script:
"""
bowtie2 -x reference_genome -U ${reads} -S aligned_${reads.getName()}
"""
}
process fastqc {
conda 'bioconda::fastqc'
publishDir 'results/fastqc'
input:
path reads
output:
path "fastqc_${reads.getName()}"
script:
"""
fastqc ${reads} -o results/fastqc
"""
}
workflow {
// Define channels
Channel.fromFilePairs(params.reads)
.ifEmpty { error "Cannot find reads!" }
.set { read_pairs_ch }
// Execute processes
align(read_pairs_ch)
fastqc(read_pairs_ch)
}
Click here for an annotated version of the mock-pipeline
```
nextflow.enable.dsl=2 // Enable Nextflow DSL2 syntax
// Define pipeline input parameters
params.reads = 'data/sample_{1,2}.fastq' // Specify the path to the input read files
// Define the align process for aligning reads to a reference genome
process align {
conda 'bioconda::bowtie2' // Specify the Conda environment for Bowtie2
publishDir 'results/aligned' // Set the output directory for aligned results
// Define the input for the process
input:
path reads // Input will be the path to the read files
// Define the output for the process
output:
path "aligned_${reads.getName()}" // Output will be the aligned reads with a specific naming pattern
// Define the script to be executed for this process
script:
"""
bowtie2 -x reference_genome -U ${reads} -S aligned_${reads.getName()} // Run Bowtie2 alignment
"""
}
// Define the fastqc process for quality control on the reads
process fastqc {
conda 'bioconda::fastqc' // Specify the Conda environment for FastQC
publishDir 'results/fastqc' // Set the output directory for FastQC results
// Define the input for the process
input:
path reads // Input will be the path to the read files
// Define the output for the process
output:
path "fastqc_${reads.getName()}" // Output will be the FastQC report with a specific naming pattern
// Define the script to be executed for this process
script:
"""
fastqc ${reads} -o results/fastqc // Run FastQC on the reads and save the output
"""
}
// Define the main workflow that orchestrates the execution of processes
workflow {
// Create a channel from the paired read files, ensuring they are correctly matched
Channel.fromFilePairs(params.reads)
.ifEmpty { error "Cannot find reads!" } // Throw an error if no reads are found
.set { read_pairs_ch } // Set the channel to read_pairs_ch for later use
// Execute the align process with the paired reads
align(read_pairs_ch)
// Execute the fastqc process with the paired reads
fastqc(read_pairs_ch)
}
```
Note
Restaurant Kitchen Analogy
Imagine you're running a restaurant, and you have a menu of dishes (the bioinformatics analyses). Each dish requires several ingredients and specific cooking processes.

Processes (Chefs/Cooking Steps)
Each process in the pipeline is like a chef in the kitchen responsible for a specific dish or part of a dish. For example, one chef (the align process) prepares the protein by marinating it and cooking it to perfection, while another chef (the fastqc process) ensures the freshness of the ingredients by performing quality checks.
Channels (Ingredient Delivery System)
The channels are like the ingredient delivery system that brings fresh ingredients to the kitchen. In our analogy, the channel takes orders (files) from suppliers (input files) and delivers them to the chefs (processes) when they are ready to cook. For instance, the channel delivers paired sample reads to both the align chef and the fastqc chef.
Workflow (Kitchen Operations/Head Chef)
The workflow is the overall management of the kitchen, orchestrating how and when each chef works. It ensures that the chefs have the ingredients they need (input channels) and specifies the order of cooking (process execution). For example, the workflow ensures that the align chef starts cooking before the fastqc chef checks the ingredients, ensuring everything is prepared in the correct sequence.
Directives (Kitchen Rules):
The directives are the kitchen rules and guidelines, like the types of equipment (Conda environments) each chef should use and how they should present the finished dishes (output directories). For instance, a directive might state that the align chef must use specific tools (bowtie2) and that the results should be plated in a particular way (published to the 'results/aligned' directory).

Image source: salmon aligner (no joke, this is their logo).
In this tutorial we are running a curated Nextflow workflow. The workflow will index the provided genome, align sequences (1/3), run QC and output a summary.
The alignment is carried out using Salmon, used for quantifying the expression levels of transcripts from RNA-Seq data. Unlike traditional aligners that map reads to a reference genome, Salmon directly estimates transcript abundance by utilizing the concept of pseudo-alignment (first mapping reads to a database of known short sequences, and then using these mapped positions to infer the position of each read on the target genome/transcriptome).
FastQC is used to do QC and MultiQC to summarize FastQC and Salmon outputs.
To execute the provided example, change directory to the nextflow_tutorial folder (cd nextflow_tutorial) and execute:
nextflow transcriptome_qc.nf
The output should be similar to this:
N E X T F L O W ~ version 24.04.4
Launching `transcriptome_qc.nf` [admiring_knuth] DSL2 - revision: 3004fdc561
R N A S E Q - N F P I P E L I N E
===================================
transcriptome: /home/jovyan/data-store/nextflow_tutorial/data/ggal/transcriptome.fa
reads : /home/jovyan/data-store/nextflow_tutorial/data/ggal/gut_{1,2}.fq
outdir : results
executor > local (4)
[e1/6abbeb] process > index [100%] 1 of 1 ✔
[f4/d72072] process > fastqc (FASTQC on gut) [100%] 1 of 1 ✔
[24/30d512] process > quantification (1) [100%] 1 of 1 ✔
[98/1c93ee] process > multiqc (1) [100%] 1 of 1 ✔
nextflow.enable.dsl=2
- Enables DSL2: we are enabling Nextflow's second domain-specific language (DSL2), which allows for more structured and modular workflows.
params.reads = "$baseDir/data/ggal/gut_{1,2}.fq"
params.transcriptome = "$baseDir/data/ggal/transcriptome.fa"
params.multiqc = "$baseDir/multiqc"
params.outdir = "results"
- These lines define input parameters for the workflow, specifying file paths for the RNA-Seq reads and the transcriptome.
println """\
R N A S E Q - N F P I P E L I N E
===================================
transcriptome: ${params.transcriptome}
reads : ${params.reads}
outdir : ${params.outdir}
"""
- This block prints the title and input parameters to the console, providing immediate feedback about the configuration.
transcriptome_file = file(params.transcriptome)
- This line creates a file object for the transcriptome, making it easier to reference later in the script.
process index {
conda 'bioconda::salmon'
input:
path transcriptome_file
output:
path 'index'
script:
"""
salmon index --threads ${task.cpus} -t ${transcriptome_file} -i index
"""
}
- This is the indexing process, which creates a binary index for the transcriptome using Salmon.
- It uses a single directive (
conda 'bioconda::salmon'): it specifies that the process should run in thebioconda::salmonenvironment (already present). - Input and Output: It takes the transcriptome as input and produces an output named
index.
Channel.fromFilePairs(params.reads)
.ifEmpty { error "Cannot find any reads matching: ${params.reads}" }
.set { read_pairs_ch }
- This block creates a channel from paired FASTQ files based on the specified pattern and checks for the existence of files.
process quantification {
conda 'bioconda::salmon'
input:
path index
tuple val(pair_id), path(reads)
output:
path "${pair_id}_quant"
script:
"""
salmon quant --threads ${task.cpus} --libType=U -i ${index} -1 ${reads[0]} -2 ${reads[1]} -o ${pair_id}_quant
"""
}
- This process quantifies the expression levels of transcripts using the Salmon quantification tool (
salmon quant). - As input takes the index created in the index process and paired reads; as output it produces quantification results named after the sample IDs.
process fastqc {
conda 'bioconda::fastqc'
tag "FASTQC on ${sample_id}"
input:
tuple val(sample_id), path(reads)
output:
path "fastqc_${sample_id}_logs"
script:
"""
mkdir fastqc_${sample_id}_logs
fastqc -o fastqc_${sample_id}_logs -f fastq -q ${reads}
"""
}
- This process runs FastQC on the input reads to assess their qualit and generates a log directory for the FastQC results.
process multiqc {
publishDir params.outdir, mode: 'copy'
conda "bioconda::multiqc"
input:
path fastqc_files
path quant_files
output:
path 'multiqc_report.html'
script:
"""
multiqc .
"""
}
- This process runs MultiQC to aggregate the results from FastQC and Salmon quantification and produces an the MultiQC HTML report summarizing the results.
workflow {
index_ch = index(transcriptome_file)
fastqc_ch = fastqc(read_pairs_ch)
quant_ch = quantification(index_ch, read_pairs_ch)
multiqc(fastqc_ch, quant_ch)
}
- The workflow block defines the order of execution for the processes, linking the outputs of one process to the inputs of another, Establishing the flow of data throughout the pipeline.
We can now visualize the output, in results.

Image source: wikipedia.
This is BY NO MEANS something simple to grasp. If anything, only a fool would try to explain Nextflow in 1 hour (I am, indeed, that fool).
Nextflow is complex, and it has a steep learning curve. There are things we did not cover:
- What's the
workfolder created? - How can I check logs?
- Error handling??!?
- What's a
resume?
Learning Nextflow is, however, extremely rewarding:
- Extremely well integrated with containers.
- Support for SLURM and PBS HPC systems.
- "Easily" sharable workflows.
- Honestly, pretty good on your CV.
Here are some great Nextflow resources for you to learn more:
- General Documentation
- Nextflow training
- Find well supported and extremely powerful pipelines at nf-co.re
- rna-seq specific pipeline using the aligner of your choice
- The full extended tutorial this was inspired by (note: this might only run on previous iterations of Nextflow, using dsl1)
- CyVerse Webinar on NextFlow (shoutout to Conner Copeland!)