Applications of AI ML in Bioinformatics - ua-datalab/Bioinformatics GitHub Wiki

Important
🕐 Schedule
- 3:00pm-3:10pm: Welcome and introduction to the topic
- 3:10pm-3:40pm: What is Machine Learning (and how can we apply it in biology/bioinformatics?)
- 3:40pm-4:00pm: Real world applications of AI/ML in Bioinformatics (tools and applications)
Important
✅ Expected Outcomes
- Understanding what Machine Learning is
- Exposure to topics of interest
Modern biology heavily relies on data from advanced techniques. For precision medicine to be effective, we need to properly analyze this data. Tools from bioinformatics and artificial intelligence are essential in transforming big data into actionable information.
- Information science focuses on how to collect, organize, and share information, covering areas like library science and data management, with uses in education and business.
- Artificial intelligence (AI) mimics human thinking in machines, performing tasks like speech recognition and decision-making, and is widely used in healthcare and self-driving cars.
- Machine learning (ML), a part of AI, helps computers learn from data to make predictions without direct programming, applied in areas like protein structure and disease diagnosis.
- Deep learning (DL), a type of ML, uses neural networks to process data in layers, greatly improving AI's ability to perform tasks like image and speech recognition with high accuracy.

Deep learning algorithms excel at tasks such as image and speech recognition, text generation, and robotic control. By learning complex patterns from raw data, these algorithms have significantly advanced artificial intelligence capabilities.
Click to Expand: What is Machine Learning?
Machine learning, a branch of artificial intelligence, develops algorithms that learn from data to make predictions or decisions. It's widely applied in fields like image recognition, natural language processing, autonomous vehicles, and medical diagnosis (Acosta et al., 2022).
Machine learning algorithms require numeric data, typically represented as matrices of samples and features. When data isn't numeric, feature engineering transforms it into usable numeric features (Roe et al., 2020).
High-quality, accurate, and representative data is crucial for machine learning algorithms to learn correct patterns and make accurate predictions (Habehh and Gohel, 2021). Expert-crafted features, designed by domain specialists, can enhance algorithm performance, especially with limited training data (Lin et al., 2020).
Common machine learning algorithms include (Hastie et al., 2009; Jovel and Greiner, 2021):
- Linear regression: Finds best-fit lines for variable relationships
- Logistic regression: Predicts event probabilities
- Decision trees: Uses binary decisions for predictions
- Random forest: Combines multiple decision trees
- Support vector machines: Finds optimal separating hyperplanes
- Neural networks: Uses interconnected nodes to learn complex patterns
In summary, machine learning is a versatile tool requiring numeric, high-quality data and sometimes expert-crafted features. Various algorithms are available to address different problems.
Click to Expand: What is Deep Learning?
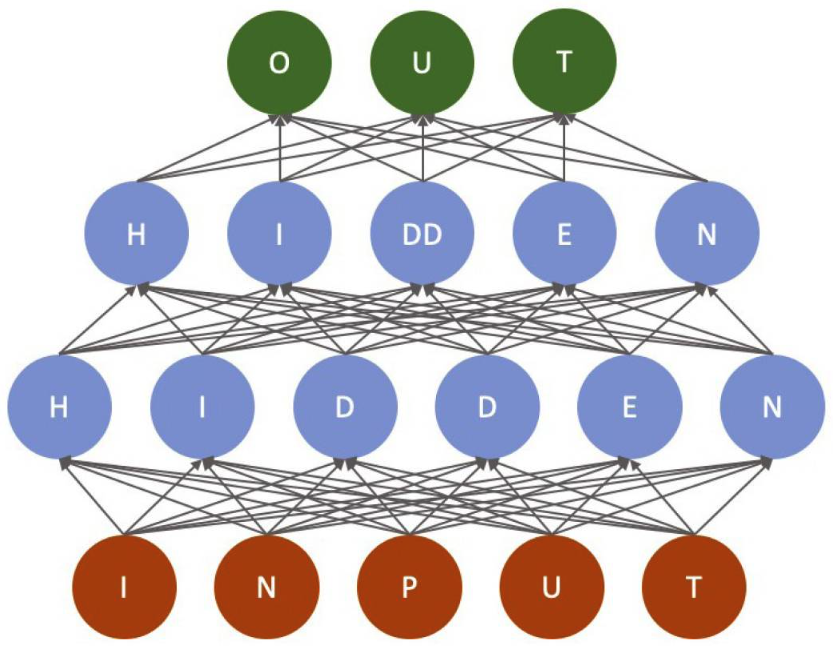
Neural networks, introduced decades ago, have evolved from simple structures to deep neural networks with multiple layers. Modern deep learning models typically consist of input, hidden, and output layers, leveraging increased computational power to handle more complex architectures.
The input layer receives raw data or features, with each node representing a data point. Hidden layers transform this data into abstract representations, with multiple layers defining the "depth" of the network. The output layer produces the final prediction based on the processed information.
Deep learning models are trained by adjusting connection weights to minimize errors, often using large labeled datasets and optimization techniques like backpropagation and gradient descent. Model architecture is typically chosen empirically for each specific task.
Unlike traditional machine learning, deep learning automatically detects higher-level features. This can lead to reduced explainability, which is particularly important in fields like healthcare where understanding decision-making processes is crucial (Sarker, 2021; the Precise4Q consortium et al., 2020).
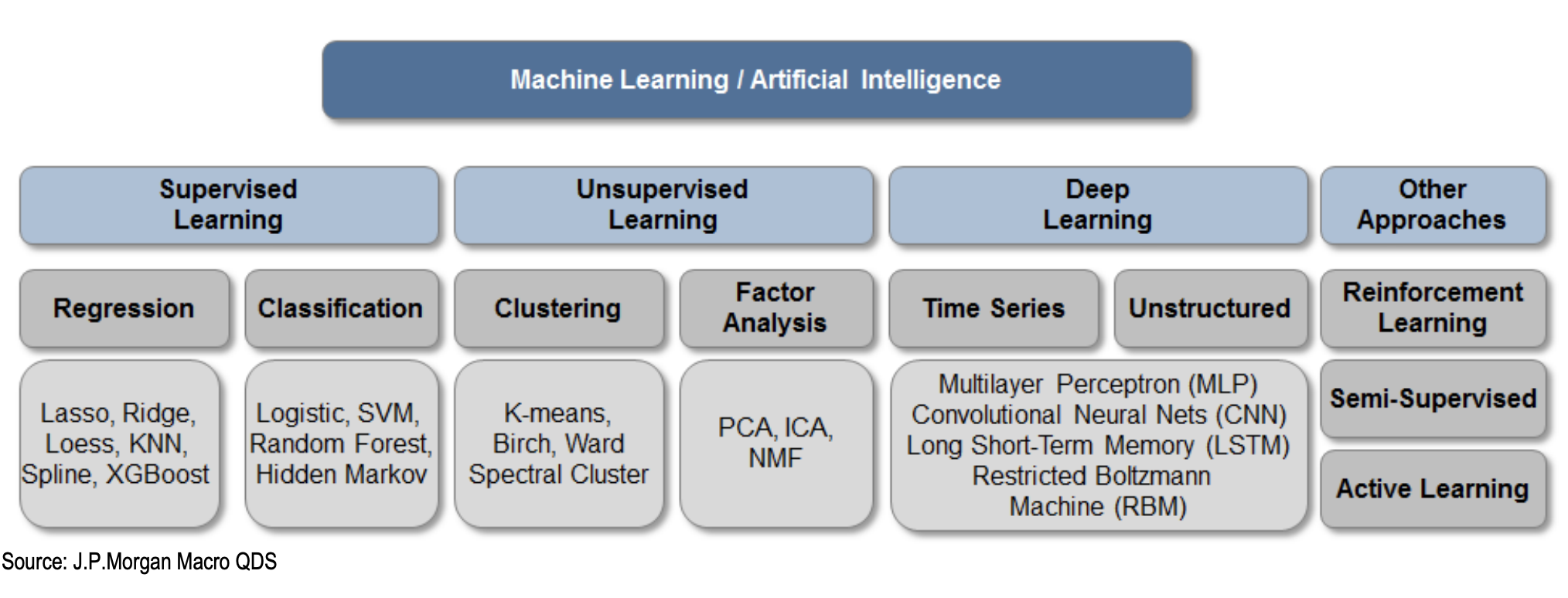
Differences between machine learning using traditional algorithms and machine learning using deep neural networks.
| ML | DL | |
|---|---|---|
| Algorithms | Many different (SVM, Decision Trees, kNN, ...) | Defined by architecture (RNN, GAN, LSTM, ...) |
| Data size | Can work well with smaller inputs | Requires large amount of data |
| Performance | Typically extremely fast | Computational complexity depends on the architecture |
| Features | Hand-crafted | Can be learned |
| Preprocessing | Significant effort | Can be trained on raw data |
| Fine tuning | Setting the algorithm parameters | Can be performed automatically during training |
| Complexity | Typical simple mathematical models | Depends on the architecture (highly flexible) |
| Transparency | Typically transparent | Hard to transparently show decision making |
| Explainability | Typically explainable | Hard to show the reasoning process |
We live surrounded by data. Big or small. Large or minimal. Through the power of statistics, we have learned to leverage this data to give us further insights in the data that the world around us is full of.
ML/AI has accented this data discovery, but it comes with a couple of caveats:
- ML/AI is difficult to learn (but is becoming more and more accessible!) and apply
- ML/AI requires a lot of resources in order to execute: platforms require large GPU clusters and disk space; Models require a massive amount of power to be created.
Here are a few examples of groups that were able to take this technology and give us a window on the potential applications of ML/AI in Bioinformatics.
Important
Most of the following examples are searchable through Papers With Code: a powerful website that allows to look for papers that have made their code available.
This is extremely useful for folks (and skeptics!) that want to take a look at the code itself!

This paper proposes a machine learning method for identifying differentially expressed genes (DEGs) in RNA sequencing data.
By using epigenomic data from histone modifications such as acetylation, the model learns the association of these markers with gene expression and uses this to predict DEGs. The authors found that the model using InfoGain feature selection and Logistic Regression classification was most effective for identifying DEGs in the response to ethylene, and validated the prediction results through qRT-PCR.
While RNA sequencing (RNA-seq) is a popular method for identifying differentially expressed genes (DEGs), its sensitivity can be limited by experimental design and data analysis processes. ML is being used to find ways to enhance DEG identification.
Here's how ML enhances sensitivity:
- Identifying Subtle Patterns: ML algorithms can learn complex patterns in data that traditional RNA-seq methods might overlook. This allows ML to identify DEGs that exhibit subtle changes in expression, leading to a higher sensitivity. For example, the study in the source used ML to analyze features related to histone modifications and their connection to gene expression regulation in response to ethylene.
- Integration of Multiple Data Types: ML models can integrate information from various sources, such as RNA-seq data, histone modification data, and DNA methylation data. This comprehensive approach provides a richer context for identifying DEGs, increasing sensitivity compared to using RNA-seq data alone. The study used data from histone H3 modification markers (H3K9Ac, H3K14Ac, and H3K23Ac) and RNA-seq data from Arabidopsis seedlings treated with or without ethylene gas to extract features for their ML analysis.●
- Overcoming Bias and Limitations: ML can help mitigate biases and limitations inherent in RNA-seq experimental design and data analysis. For example, ML models can account for variations in sequencing depth, library preparation, and normalization methods, leading to more accurate DEG identification. The study successfully predicted DEGs that were missed by traditional RNA-seq analysis, such as EIN3-BINDING F BOX PROTEIN 1 (EBF1).
- Prediction of Novel DEGs: By learning from existing data, ML models can predict the differential expression of genes that have not been previously identified as DEGs. This predictive capability expands the pool of potential DEGs for further investigation, increasing the sensitivity of DEG identification. The study found that more than 70% of the genes predicted by their ML model were not previously reported as DEGs in response to ethylene.
The study was split into 2 main sections: Feature Selection (idenfitying the DEGs) and Classification (ML)
Feature Selection:
- Information Gain (InfoGain): This algorithm measures the reduction in entropy (uncertainty) of a variable given the value of another variable. It was used to identify features that were most informative for predicting DEGs.
- Correlation Feature Selection (CFS): This algorithm selects a subset of features that are highly correlated with the class (DEG status) while having low inter-correlation among themselves.
- ReliefF: This algorithm assigns weights to features based on their ability to distinguish between instances that are close to each other but belong to different classes.
Classification:
- Logistic Regression: This algorithm models the probability of a binary outcome (DEG status) based on a linear combination of input features. It was found to be the most effective classification method when combined with InfoGain feature selection. Classification Via Regression: This algorithm transforms a regression problem into a classification problem. It showed good performance in predicting DEGs.
- Random Forest: This algorithm constructs multiple decision trees during training and outputs the class that is the mode of the classes output by individual trees. It was found to be the best-performing algorithm when applied to human and rice cell data.
- Logistic Model Trees (LMT): This algorithm combines logistic regression and decision tree learning.
- Random Subspace: This algorithm creates multiple classifiers by training them on random subsets of features. It performed well in predicting DEGs.
Their research suggests that combining epigenomic information with RNA-seq data analysis using machine learning methods could significantly improve the sensitivity of DEG identification.
Large-scale machine learning-based phenotyping significantly improves genomic discovery for optic nerve head morphology
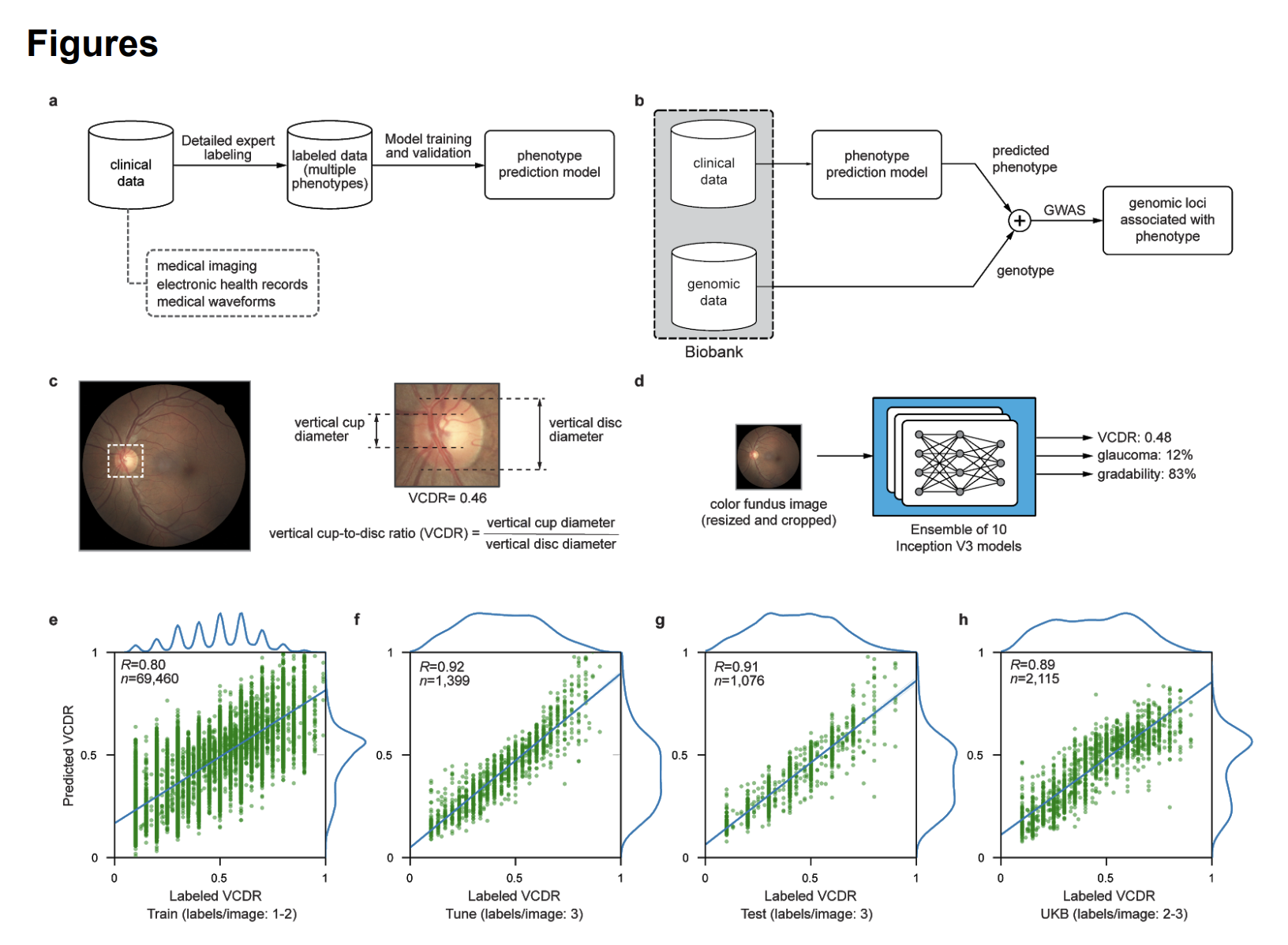
This article examines the use of machine learning (ML) models to predict glaucoma-related traits from color fundus photographs. The authors developed an ML model that accurately predicts the vertical cup-to-disc ratio (VCDR) and identified 299 independent genome-wide significant hits in 156 loci associated with VCDR, replicating known loci and discovering 92 novel ones.
This paper used machine learning (ML) to automatically predict VCDR from fundus photographs, an important endophenotype for glaucoma. The ML model was trained using 81,830 fundus photographs that were graded by experts for image gradability, VCDR, and referable glaucoma risk. These photographs were split into training, tuning, and test sets. The authors trained an ensemble of ten Inception V3 deep convolutional neural networks, a type of ML model well-suited for image analysis.
Once trained and validated, the ML model was applied to 175,337 fundus photographs from the UK Biobank. The model predicted VCDR for each image and also identified images that were ungradable.
The authors conducted a genome-wide association study (GWAS) using the ML-based VCDR predictions. They compared the results to previous GWAS of VCDR, including one based on manually-labeled UK Biobank images. The ML-based GWAS replicated a majority of known genetic associations with VCDR and discovered 92 novel loci. This finding suggests that ML-based phenotyping can improve the power of genomic discovery.
DeepLabCut (DLC) is an open-source, deep-learning-based software designed for pose estimation. It enables researchers to track and analyze movements of animals in videos by detecting specific body parts without the need for physical markers.
It was inspired by the need for precise, automated tracking of animal behavior in neuroscience and behavioral research. The software is built upon DeepLab (Chen et al., 2016), a deep-learning framework initially designed for image segmentation, utilizing ResNet (Residual Networks) as the backbone architecture. It employs transfer learning from pre-trained models on large image datasets, refining them with user-labeled frames for precise keypoint detection. DLC applies this framework to the problem of keypoint detection, allowing researchers to track body parts efficiently. viso.ai has a quick rundown on what DeepLab is.
DeepLabCut is widely used across various scientific fields, including:
- Neuroscience – Tracking animal movements to study motor functions and behavioral responses.
- Biomechanics – Analyzing motion and gait in animals and humans.
- Ecology & Ethology – Studying natural behaviors of animals in the wild or laboratory settings.
- Medical & Rehabilitation Research – Assessing motor impairments in preclinical models of neurological disorders.
- Sports Science– Analyzing human motion for performance improvement and injury prevention.
- Human-Computer Interaction (HCI) – Tracking human hand and facial movements for gesture recognition.

SLEAP (Social LEAP Estimates Animal Poses) is an open-source deep-learning-based tool for multi-animal pose estimation. It is designed as an alternative to DeepLabCut (DLC) but focuses on handling multiple interacting animals more effectively.
In order to help tracking multiple objects, SLEAP uses a similar setup as DLC but also includes multiple network architectures for different use cases:
- Top-down models: first detecting the entire animal and then estimating its pose.
- Bottom-up models: directly detecting individual keypoints across all animals.
- Fully convolutional networks for segmentation-based approaches.
- ResNet and HRNet architectures for feature extraction and pose estimation.

Important
Who won the Nobel Prize for chemistry in 2024? One of the winners is Demis Hassabis and John Jumper, from the team that developed AlphaFold for their work on “protein structure prediction”.
AlphaFold is an artificial intelligence (AI) system developed by DeepMind for predicting the 3D structure of proteins from their amino acid sequences. It has revolutionized structural biology by providing highly accurate protein structure predictions, which are crucial for understanding biological processes, drug design, and many other applications in molecular biology.
How AplhaFold works:
- Input: The primary input to AlphaFold is a protein’s amino acid sequence. This is analogous to a string of letters representing the order of the protein's building blocks.
- Multiple Sequence Alignment (MSA): AlphaFold compares the input sequence with known protein sequences in public databases. This comparison, called a multiple sequence alignment (MSA), helps AlphaFold understand evolutionary relationships between proteins and infer constraints on how the protein can fold.
- Neural Network Architecture: AlphaFold uses a transformer neural network model, which processes both the sequence data and the information from the MSA to make predictions about how close different pairs of amino acids will be in the 3D structure. It also models the geometric constraints of protein folding, like bond angles and distances between atoms.
- Structure Prediction: The model generates a predicted 3D structure, which it refines iteratively. AlphaFold optimizes the prediction by minimizing errors related to the physical and chemical properties of proteins.
- Confidence Metric: Along with the 3D structure, AlphaFold provides a confidence score (called a pLDDT score), which indicates how certain the model is about each part of the structure.
![]()
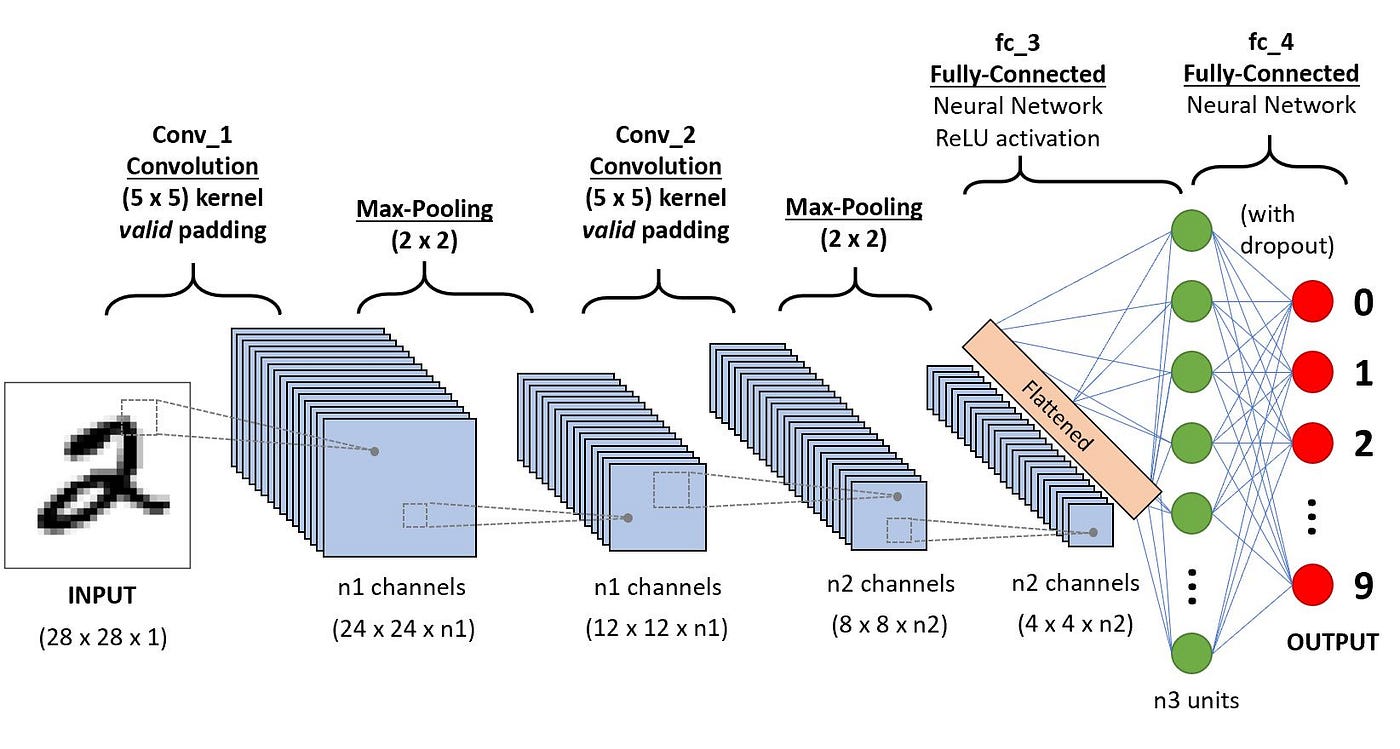
DeepVariant is an open-source software tool developed by Google for variant calling in genomics. It uses deep learning techniques to identify genetic variants from next-generation sequencing (NGS) data. DeepVariant processes the data through a neural network, transforming raw sequence data (from a BAM file or CRAM) into images (a 2 dimensional matrix) of aligned reads and then classifies these images to predict the presence of genetic variants, such as SNPs (single nucleotide polymorphisms) and indels (insertions or deletions).
In order to carry out the analysis of the matrices, DeepVariant uses a Convolutional Neural Network (CNN), a method that is often used in image analysis, to the pileup of images. The CNN then analyzes the pileup images to detect patterns associated with genetic variants, learning from both variant and non-variant regions.
Why DeepVariant is Effective:
- Deep Learning for Accuracy: Unlike traditional rule-based variant callers (like GATK), DeepVariant uses deep learning to learn subtle patterns in sequencing data, which helps it to be more accurate, particularly in challenging genomic regions (e.g., repetitive sequences, homopolymers).
- Training on Large Datasets: DeepVariant was trained on large reference datasets such as the Genome in a Bottle (GIAB) benchmarks, improving its ability to generalize and accurately call variants in various samples.
How is it so affective?
- DeepVariant creates tensors from the pileup images. A tensor is a generalization of vectors and matrices to potentially higher dimensions, and it's a fundamental data structure used in deep learning models. Tensors are used to represent data in a way that can be efficiently processed by neural networks.
- In DeepVariant, a pileup image tensor is a 3D data structure. It holds sequencing information in a format that a deep learning model can process:
- The first dimension is typically the height (number of reads aligned at a particular position).
- The second dimension is the width (corresponding to base positions along the reference genome).
- The third dimension contains different channels representing additional features (e.g., base information, quality scores, mapping quality, strand information).
- This tensor then becomes the input to the CNN, which then the CNN interprets the tensor’s patterns to classify positions in the genome as variant or non-variant.
Updated: 03/26/2025 (M. Cosi)
UArizona Data Lab, Data Science Institute, University of Arizona.

