PeakCallAndMDA - suimye/NGS_handson2015 GitHub Wiki
3. Peak Callに基づくタンパク質-DNA結合領域の検出
今回の実習では、Biolinux8で利用できるMACS14を利用します。 ノイズフィルタリングを行った前と後のデータでpeak callの結果を比較しましょう!!!
3.1 早速 MACSをかけてみる
#ノイズフィルタリング後のデータ
macs14 -t sample.uq.drm.rmsk.bam --name=sample -c input.uq.drm.rmsk.bam -f BAM -g hs -- wig
#ノイズフィルタリング前のデータでも行い、比較してみよう!
macs14 -t sample.bam --name=sample_before -c input.bam -f BAM -g hs -- wig
出力
iu@bio[tutorial150806] macs14 -t sample.uq.bam --name=sample -c input.uq.bam -f BAM -g hs -- wig
INFO @ Tue, 04 Aug 2015 00:49:18:
# ARGUMENTS LIST:
# name = sample
# format = BAM
# ChIP-seq file = sample.uq.bam
# control file = input.uq.bam
# effective genome size = 2.70e+09
# band width = 300
# model fold = 10,30
# pvalue cutoff = 1.00e-05
# Large dataset will be scaled towards smaller dataset.
# Range for calculating regional lambda is: 1000 bps and 10000 bps
INFO @ Tue, 04 Aug 2015 00:49:18: #1 read tag files...
INFO @ Tue, 04 Aug 2015 00:49:18: #1 read treatment tags...
INFO @ Tue, 04 Aug 2015 00:49:18: tag size: 36
INFO @ Tue, 04 Aug 2015 00:49:23: 1000000
INFO @ Tue, 04 Aug 2015 00:49:27: #1.2 read input tags...
INFO @ Tue, 04 Aug 2015 00:49:31: #1 tag size is determined as 36 bps
INFO @ Tue, 04 Aug 2015 00:49:31: #1 tag size = 36
INFO @ Tue, 04 Aug 2015 00:49:31: #1 total tags in treatment:
..... 中略.....
INFO @ Tue, 04 Aug 2015 00:50:08: #5 Done! Check the output files!
MACSの解析結果のファイル MACSの出力の説明について
-
peaks.xls
- chr 染色体番号
- start ピークの開始
- end ピークの終了位置
- length ピーク領域の長さ
- summit ピークの頂点の開始位置からの相対位置
- tags ピーク領域のタグ数
- -10*LOG10(pvalue)
- fold_enrichment 基準からの倍差
- FDR(%) %表示したFDR値
-
negative_peaks.xls
- ctrl側のピークの情報
-
peaks.bed
- 染色体番号
- 開始座標
- 終了座標
- Peak ID
- スコア
bedファイルはIGVでのpeak位置確認や、その他の解析のために整形・利用できるので便利。
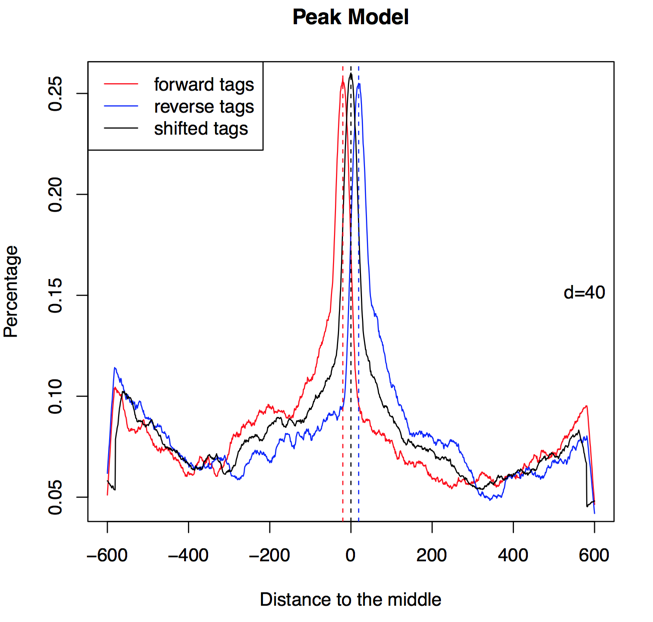
3.2 Rを使ってMACSで行ったtag-shift peakのモデルの具合をcheckする
R --vanilla < sample_before_model.r
Peakを拾って、詳細な解析のための下準備をする
今回はscoreの高い順番に50 peakを取得
cat sample_peaks.bed |sort -k5 -n -r >sample_peaks.sort.bed
headで確認して見ると
head sample_peaks.sort.bed
chr17 25301798 25304698 MACS_peak_662 3100.00
chr17 25287092 25288357 MACS_peak_648 2425.18
chr17 25268055 25269504 MACS_peak_632 2202.03
chr17 25285964 25286720 MACS_peak_647 1622.51
chr17 81181350 81182196 MACS_peak_2505 1606.45
chr17 51183091 51183752 MACS_peak_1618 1515.69
chr17 25288564 25289503 MACS_peak_649 1464.25
chr17 22021833 22022232 MACS_peak_594 1229.63
chr17 22023645 22023897 MACS_peak_599 1055.50
chr17 22022568 22022976 MACS_peak_596 986.64
top50を取得
head -n50 sample_peaks.sort.bed >top50.bed
タンパク質-DNA結合領域(Peak)の配列解析
- IGVを使って、個々にしらべる。
- top100を使って、motif discoveryをする。
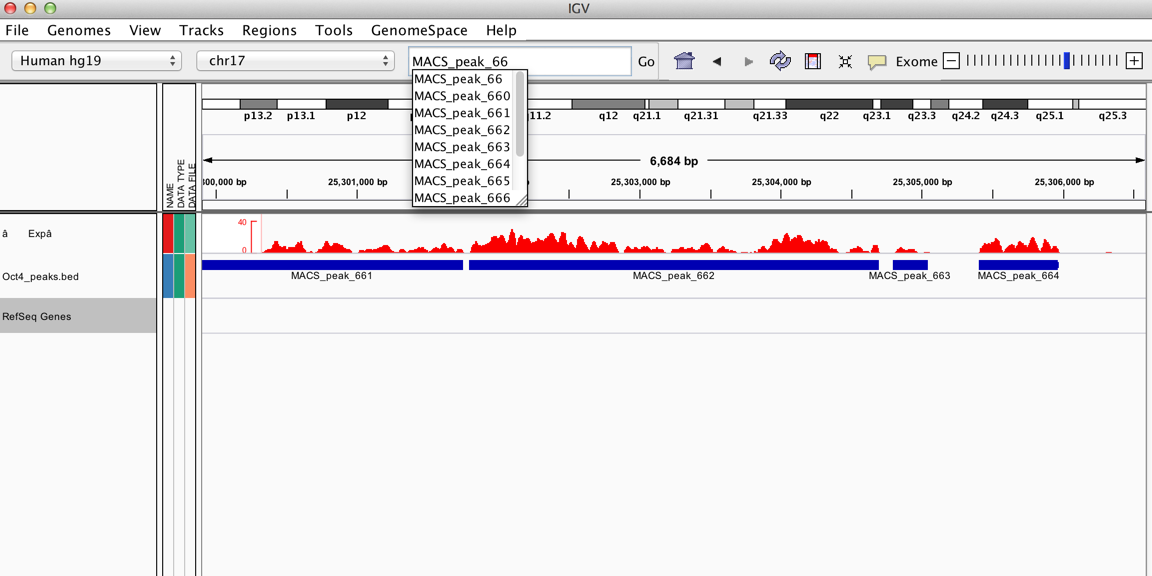
4.1 champion peakのTFBS searchをやってみよう
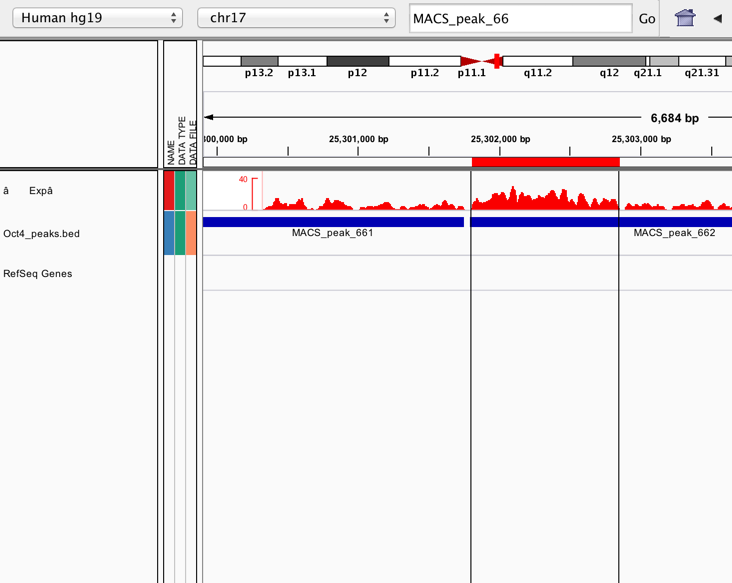
- どうやら、MA_662がNo.1らしい。
- IGVでMACS_peak_662をsearch.
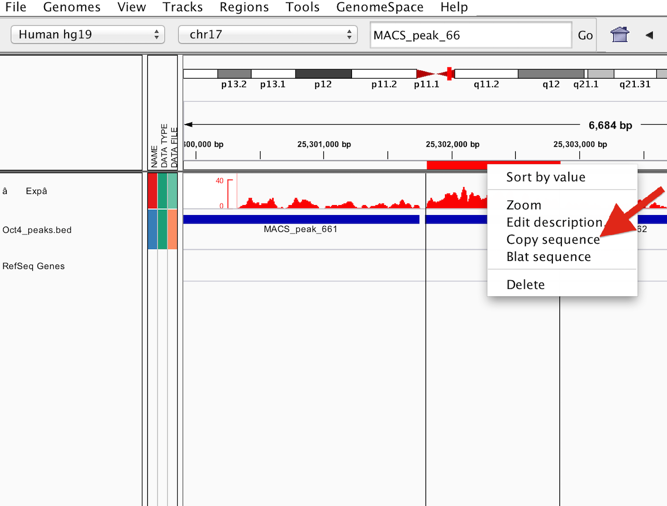
- 目的位置を赤いバー囲むようにクリックで選択する
- 赤いバー右クリック、peakの領域の塩基配列を取得する
TFBS search (簡易版、検索する転写因子が決まっている場合)
- physbinderにアクセス
- Use Methodを選択。
- コピーした配列をペーストする。
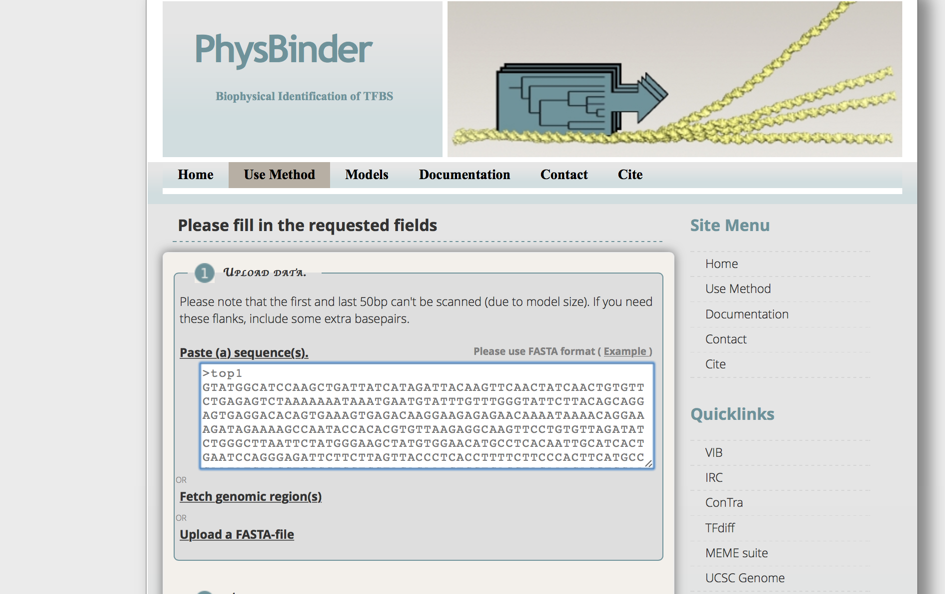
-
fasta形式になるように、先頭に">文字列"をつける。
-
TFBS modelを選択する。
解析中の画面
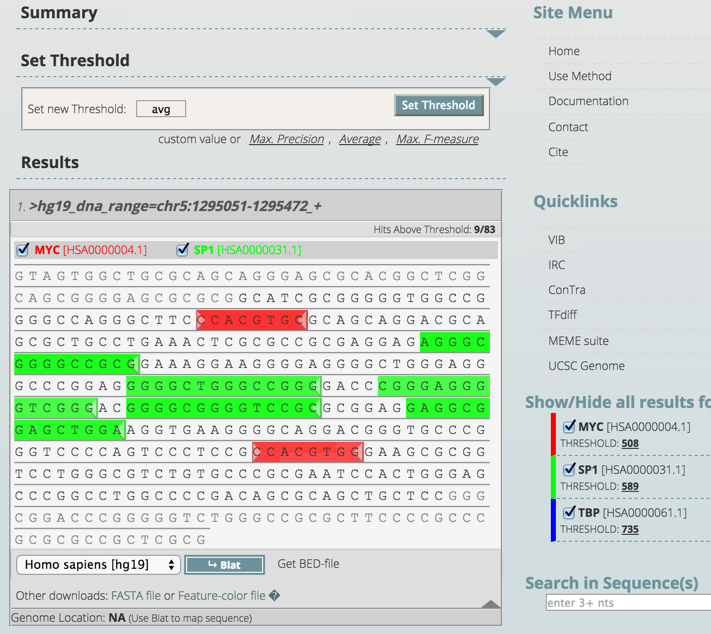
解析結果の画面
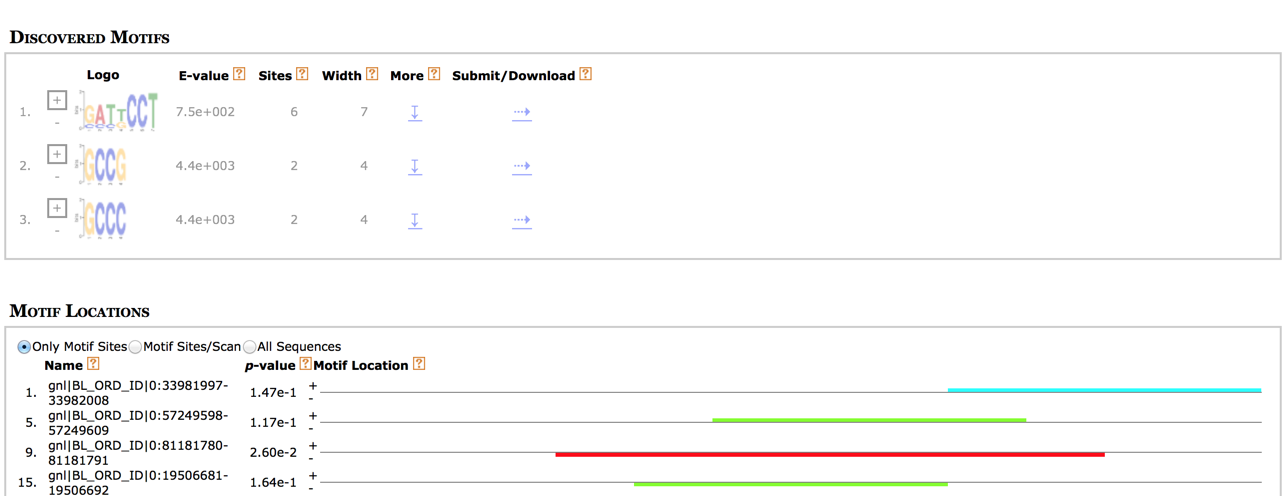
4.2 motif discovery analysis
motif discovery analysisでは、得られたDNA配列からTFBS searchよりも発見的に配列の規則を見つけ出す手法
- Peak Callで得られたbedファイルから、fastaファイルを作成する
- fastaファイルをmulti fastaにする
- MEME-ChIPを使って、motif探索を行う
databaseの作成
makeblastdb -in hg19_chr17.fa -out hg19_chr17.fa -dbtype nucl -parse_seqids
fastaファイルを作るための整形
#summitは、領域が小さいので、前後20bpほど追加する。
cat top50.bed |perl -e 'while(<>){my($chr,$st,$en,$id,$score)=split/\t/; $st=$st -10; $en=$en +10; print "$chr\t$st\t$en\t$id\t$score";}' >top50.plus.bed
#top50のdataからdata rangeを使って、blastdbcmdのコマンドを作成する。
cat top50.plus.bed|cut -f1,2,3,4|sed 's/\t/-/g'|sed 's/chr17-/blastdbcmd -db hg19_chr17.fa -entry all -range /' |sed 's/-MACS_/ >/'>top50fasta.sh
#shell scriptを実行
sh top50.fasta.sh
出力結果のcheck
iu@bio[tutorial150806] cat peak_1044 [ 1:24午前]
>gnl|BL_ORD_ID|0:33981997-33982008 chr17
TAAGATTTGCCG
fastaファイルをひとまとめにして、multi fastaを作成する
#mfa作成
iu@bio[tutorial150806] cat peak_* >top50.mfa [ 1:28午前]
#中身の確認
iu@bio[tutorial150806] head top50.mfa [ 1:29午前]
>gnl|BL_ORD_ID|0:33981997-33982008 chr17
TAAGATTTGCCG
>gnl|BL_ORD_ID|0:41231735-41231746 chr17
GTGAGAACCAAT
>gnl|BL_ORD_ID|0:51183370-51183381 chr17
TCCCCTATTCTC
>gnl|BL_ORD_ID|0:57248285-57248296 chr17
AACACACAGCTC
>gnl|BL_ORD_ID|0:57249598-57249609 chr17
GTTCCGCCCCAG
#必要のない個々のfastaファイルを削除する
iu@bio[tutorial150806] rm -f peak_* [ 1:29午前]
MEME-ChIP
MEME-ChIPへアクセスする

- 左のタブのMotif Discoveryをクリック
- MEME-ChIPを選択
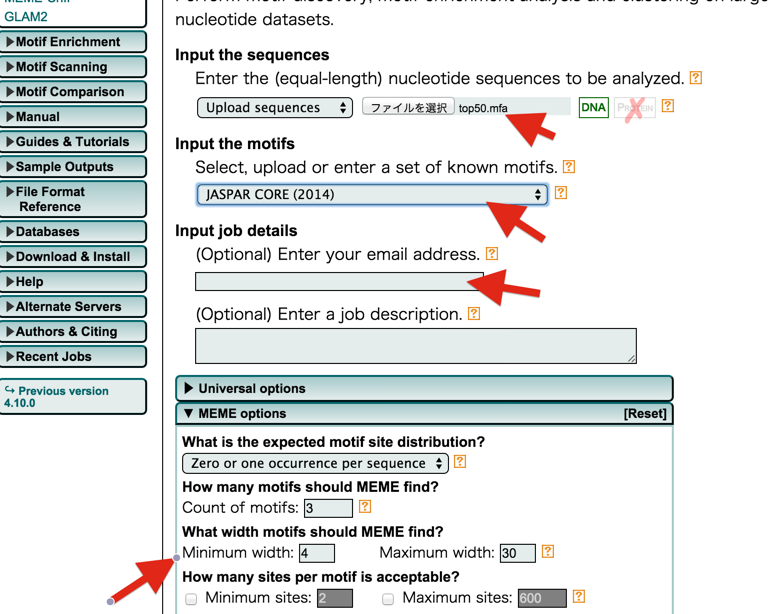
重要なパラメータについて
- How many motifs should MEME find?
- 与えた配列に対して、全体でいくつのmotifを見つけさせるか
- What width motifs should MEME find?
- motifの大きさ
- How many sites per motif is acceptable?
- 発見したmotifが利用している配列の数
結果の画面

- download、もしくはhtmlで結果をみてみる。