NGSplotsOnBiolinux8 - suimye/NGS_handson2015 GitHub Wiki
Ngsplotのbiolinux8でのinstallと解析
はじめに
NGSplotを利用する最大のオススメポイントは、アンカーとする場所(たとえば転写因子のゲノムワイドな結合部位)を指定し、その他のNGSデータのtag densityをそのアンカーから眺めることができるので、例えばpeak callの行い難いHistone修飾のChIP-seqデータなどのゲノムワイドな傾向を複数のデータを用いて俯瞰することができます。
また、アンカーは、Ngsplotの著者が用意しているtss, miRNA, ncRNA, genebodyだけではなく、オリジナルのbedファイルを利用することもできますので、NGSデータの組み合わせ解析の一つとして取り扱いやすい方法です。
doMCのパッケージがミラー先によってinstallできない印象がある。
それ以外は、大きくこけるところは無くinstallできる模様
R version 3.2.0 (2015-04-16) -- "Full of Ingredients"
installディレクトリの作成
mkdir ~/tools
mv ~/Downloads/ ~/tools
cd ~/tools
#解凍
tar xzvf ngsplot-2.47.tar.gz
pathを通す
echo 'export PATH=~/tools/ngsplot/bin:$PATH' >>~/.bashrc
echo 'export NGSPLOT=~/tools/ngsplot' >>~/.bashrc
source ~/.bashrc #PATHを有効にする。
Rのパッケージインストール
updateもすべてYesにすると10分以上かかります。
source("http://bioconductor.org/biocLite.R")
install.packages("doMC") #ミラー選択によってDLできないことがあるのでできなかった場合はミラーを変えて最トライ。
biocLite("caTools", dep=T)
install.packages("utils", dep=T) #R3.0以降はutilsは、必要ない
biocLite("ShortRead")
biocLite("BSgenome")
```
install時のwarning message
```
#BitSeqがインストールできていない状況。ただしprogramは最終的に動いた。
* DONE (GenomeInfoDb)
The downloaded source packages are in
‘/tmp/RtmpoZ1j1c/downloaded_packages’
Warning message:
In install.packages(update[instlib == l, "Package"], l, contriburl = contriburl, :
installation of package ‘BitSeq’ had non-zero exit status
```
備考:doMCが入っていないときのエラーメッセージ
```
Loading R libraries..Installing package into ‘/home/iu/R/x86_64-pc-linux-gnu-library/3.2’
(as ‘lib’ is unspecified)
Error in contrib.url(repos, type) :
trying to use CRAN without setting a mirror
Calls: install.packages -> grep -> contrib.url
In addition: Warning message:
In library(package, lib.loc = lib.loc, character.only = TRUE, logical.return = TRUE, :
there is no package called ‘doMC’
Execution halted
pathが通っているか確認
iu@bio[ctcf] ngsplotdb.py list
ID Assembly Species EnsVer NPVer InstalledFeatures
hg19 GRCh37 homo_sapiens 74.0 3.0 cgi,exon,genebody,tss,tes
ngsplotにはデフォルトでhg19のゲノム情報が入っている。これを用いて今回のデータを解析してみる。
ngs.plot.r -G hg19 -R genebody -C sample.uq.sort.bam -O CTCF.genebody -T CTCF -L 3000 -FL 200
ngs.plot.r -G hg19 -R genebody -C input.uq.sort.bam -O input.genebody -T input -L 3000 -FL 200
- -G: ゲノム(専用のデータがGDriveに落ちてる)
- -R: 解析region、いろいろ選べる。
- -C: 解析対象のbamファイル
- -O: outputフォルダの名前
- -T: plotの中のレジェンドにつく名前
- -L: 指定した領域(例えばgene bodyやTSS)の前後から何bp付け足すか。x軸の領域が決まる。
- -FL:
カバレッジの計算する際のbinの大きさFLはフラグメント (Insert)サイズ(3'側に付け足すタグ伸長ため)を入力するところです。 - -N: Nに数字を指定した場合、 N x FLが上記のLになる。
160513追記 -Dオプション
- -D: Gene database: ensembl(default), refseq
備考: sortされていないデータを用いたときのエラーメッセージ
[bam_index_build2] fail to index the BAM file.
Error in FUN(X[i](/suimye/NGS_handson2015/wiki/i), ...) : failed to build index
file: sample.uq.bam
Calls: headerIndexBam ... indexBam -> sapply -> sapply -> lapply -> FUN -> .Call
Execution halted
解析成功時のmessage
iu@bio[ctcf] ngs.plot.r -G hg19 -R genebody -C sample.uq.sort.bam -O CTCF.genebody -T CTCF -L 3000 -FL 200
Configuring variables...
Using database:
/home/iu/tool/ngsplot/database/hg19/hg19.ensembl.genebody.protein_coding.RData
Done
Loading R libraries.....Done
Analyze bam files and calculate coverageWarning message:
'isNotPrimaryRead' is deprecated.
Use 'isSecondaryAlignment' instead.
See help("Deprecated")
...........................................................................................................................................................................................................Done
Plotting figures...Done
Saving results...Done
Wrapping results up...Done
All done. Cheers!
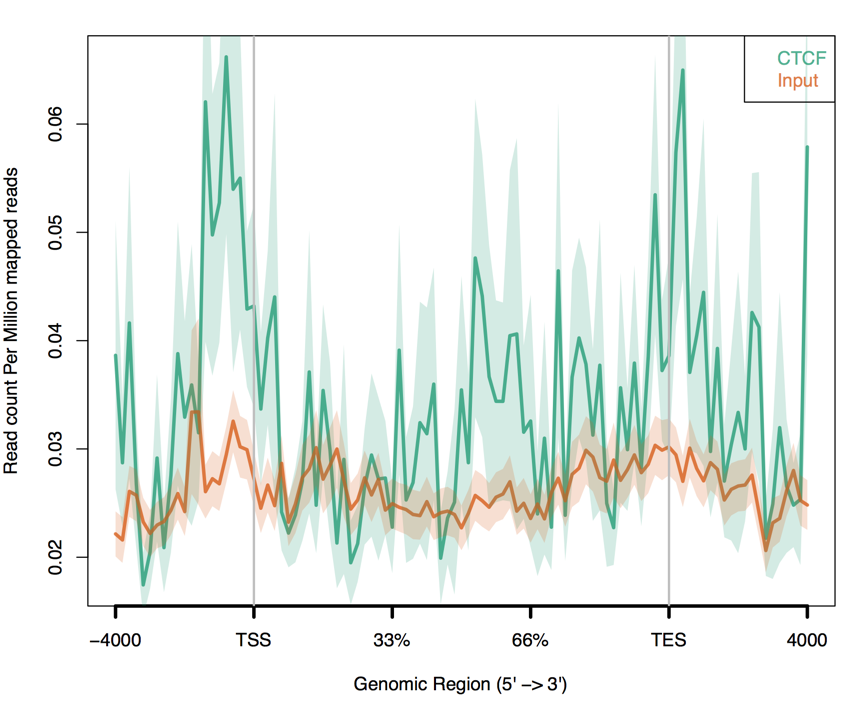
解析結果を比較してみよう
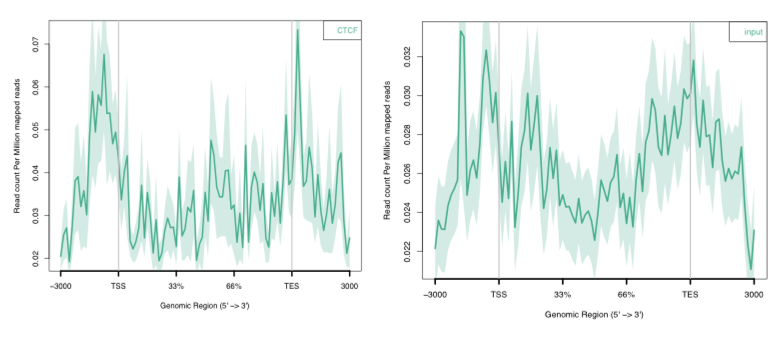
比較しやすいように同じplotに2つ描く方法
対象となるデータのリスト(list.txt)を作成し、オプションーCで読み込む。
##ファイルリストを作成する
echo 'sample.uq.sort.bam -1 "CTCF"'>list.txt
echo 'input.uq.sort.bam -1 "Input"'>>list.txt
##ngs.plotを実行
ngs.plot.r -G hg19 -R genebody -C list.txt -O CTCFandCtrlgenebody -L 4000 -FL 200
アンカーデータを指定する方法
TSSやgenebodyだけでは面白くない。そんなあなたには、オリジナルのアンカーデータをbed fileで与える方法もあります。とても簡単で、アンカーファイルとして、bed形式のデータがあればOKです。たとえば、MACSで得られるsummitファイルを中心とすれば、MACSにかけたChIP-seqデータの転写因子結合中心と、他のNGSデータ(ヒストン修飾)のゲノム領域などをその距離の相関として見る事ができるはずです。
例:anchor.bedファイルを準備した場合
- "-E"でanchor.bedファイルを指定します。
- CTCFのtag分布をanchor周りのデータを計算します。
ngs.plot.r -G hg19 -R bed -E anchor.bed -C ~/CTCF.sort.bam -O CTCF.distribution -T CTCF -L 3000 -FL 100