NGSデータの準備とmapping - suimye/NGS_handson2015 GitHub Wiki
実習環境の準備
あらかじめ解析データを入れるフォルダを作成しましょう。
# 実習フォルダの作成
mkdir ~/tutorial150806
# フォルダへ移動
cd ~/tutorial150806
##必要なデータのダウンロード
今回の実習に利用するデータをダウンロードしましょう。 google drive,dropboxに置いてあります。 講義中にDLしておいてください。
データ移動させ解凍しておく
ダウンロードできたかどうかは、ブラウザの右上にあるDownloadsをクリックすると確認できます。ダウンロード完了後、以下のコマンドを実行して解凍してください。
#ヒトゲノム配列の解凍(hg19の染色体17番のデータ)
mv ~/Downloads/hg19_chr17.zip ./
unzip hg19_chr17.zip
mv hg19_chr17/* ./
mv ~/Downloads/sample.fastq.gz ./
gzip -d sample.fastq.gz
mv ~/Downloads/nazo.fastq.gz ./
gzip -d nazo.fastq.gz
0. 今回のデータの取得とクオリティcheck
fastqファイルを取得後に、中身のデータをターミナル画面で確認する(head commandの復習: 中身をheadコマンドで確認してみる)。
head sample.fastq
@SRR445816.18948743 HWUSI-EAS366_145:3:39:5673:1812/1
ACTCCTGCCTCAGGTGATCCATCCGCCTCAGCCTCT
+
BDGGG@GDGGBG@DDE@ABFGGGB<CFEAE<:???=
@SRR445816.28703832 HWUSI-EAS366_145:3:61:8637:6699/1
ACTGACTCAAATGTTAATCTCCTTTGGCAACACTCT
+
IIIIIIIIIIIIIIIIIIIIIIEIIIIIIIIIIIII
@SRR445816.43211059 HWUSI-EAS366_145:3:94:3572:7097/1
TAAGCCCCTTCTCTTAGGATTTATAACCTCATCACT
Phred scoreに基づくsequencing qualityのチェックをしてみよう
fastqcコマンドを使って、クオリティをcheckします。出力先のディレクトリを作成した後、fastqcを行います(出力ディレクトリーには、日付を入れよう!)。
mkdir sample_qc1/
fastqc -o sample_qc1_210305/ sample.fastq
mkdir input_qc1/
fastqc -o input_qc1_210305/ input.fastq
fastqx_toolkitを使ったQCフィルタリング
QV>=20以上の塩基が80%あるreadを残すフィルタリングを実施する。
fastq_quality_filter -q 20 -p 80 -Q33 -i sample.fastq -o sample.q1.fastq
fastq_quality_filter -q 20 -p 80 -Q33 -i input.fastq -o input.q1.fastq
先ほどと同様にfastqcによる品質評価を行い、filtering前とfiltering後を比較してみてください。
mkdir sample_q1_qcout/
mkdir input_q1_qcout/
fastqc -o sample_q1_qcout/ sample.q1.fastq
fastqc -o input_q1_qcout/ input.q1.fastq
結果のHTMLファイルをクリックすると品質評価の図をみることができる。
-
リード長に伴うシーケンシングスコアの推移の比較
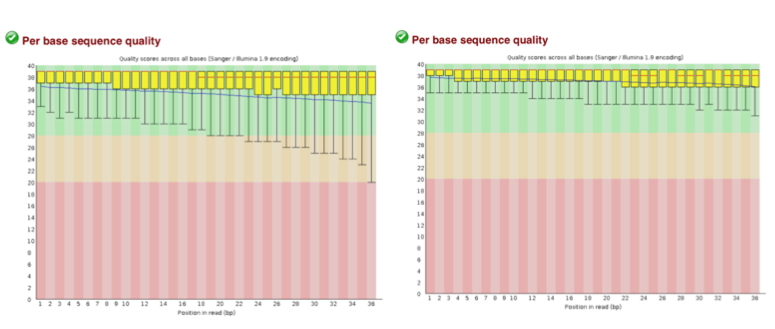
-
塩基バイアスの調査(トリムの必要あるか)
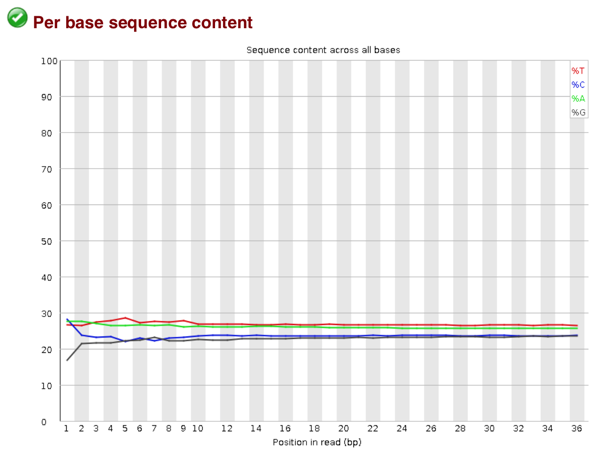
1. bowtie2を使ったゲノムへのマッピング
bowtie2 -p4 -N0 -x ./hg19_chr17/hg19_chr17 sample.q1.fastq > sample.sam
bowtie2 -p4 -N0 -x ./hg19_chr17/hg19_chr17 input.q1.fastq > input.sam
重要なオプションの解説
-x: 参照するゲノムを指定。bowtie2用のインデックス化されたゲノムが必要
-q: 入力配列。fastq形式(.fqか.fastq)
-S: アライメント後の出力ファイル名の指定
-p: 利用するスレッド数
--un: ゲノムにアライメントされなかったfastqファイルを出力するファイル
-N: アライメントの許容するミスマッチ数
-5: 5'からトリムする場合の塩基数
-3: 3'からトリムする場合の塩基数
--qc-filter: fastqファイルのPhred Scoreに基づくfiltering (先にqc filteringしていない場合に用いる)
-r: pair-end readのアライメントを行う時に
出力結果
iu@bio[tutorial150806] bowtie2 -p4 -N0 -x ./hg19_chr17/hg19_chr17 sample.q1.fastq --un sample.un.out > sample.sam
6164507 reads; of these:
6164507 (100.00%) were unpaired; of these:
0 (0.00%) aligned 0 times
1550235 (25.15%) aligned exactly 1 time
4614272 (74.85%) aligned >1 times
100.00% overall alignment rate
裏課題
暇なひと(上級者)は、sample.un.outのデータからどんな塩基配列がmapされなかったか調べてみよう。
- softwareを使う
- 地味にやる場合
fastaファイルを生成して、各種相同性検索のプログラムを実行する
wget "https://raw.githubusercontent.com/jimhester/fasta_utilities/master/scripts/fastq2fasta.pl"
- GGGenomeを使う(http://gggenome.dbcls.jp/ja/)
- 相同性検索をかける
mapping結果であるsamファイルの中身を覗いてみてみよう。
# 先頭から五行表示する
iu@bio[tutorial150806] head -n5 sample.sam [12:12午前]
@HD VN:1.0 SO:unsorted
@SQ SN:chr17 LN:81195210
@PG ID:bowtie2 PN:bowtie2 VN:2.2.4 CL:"/usr/bin/../lib/bowtie2/bin/bowtie2-align-s --wrapper basic-0 -p4 -N0 -x ./hg19_chr17/hg19_chr17 sample.q1.fastq"
SRR445816.15448471 0 chr17 5102645 1 36M *0 0 CTCCCAGGCTGGAGTGCAGTGGTGCAATCTTGGCTC HHHHHHHHHHHHHGHHHHFGGGCGGHHHHHHHHHHH AS:i:0 XS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:36 YT:Z:UU
SRR445816.28703832 0 chr17 10949619 1 36M * 0 0 ACTGACTCAAATGTTAATCTCCTTTGGCAACACTCT IIIIIIIIIIIIIIIIIIIIIIEIIIIIIIIIIIII AS:i:-6 XS:i:-6 XN:i:0 XM:i:1 XO:i:0 XG:i:0 NM:i:1 MD:Z:33C2 YT:Z:UU
iu@bio[tutorial150806]
The SAMファイルの情報一覧:
- Read Name
- SAM flag
- chromosome (if read is has no alignment, there will be a "*" here)
- position (1-based index, "left end of read")
- MAPQ (マッピングクオリティ, 0=non-unique, >10 probably unique)
- CIGAR string (insertion (i.e. introns)がリードの中にあるか否かなどに重要。例えば、splicing junctionの決定などにも利用)
- Name of mate (mate pair information for paired-end sequencing, often "=")
- Position of mate (mate pair information)
- Template length (always zero for me)
- Read Sequence
- Read Quality
- Program specific Flags (i.e. HI:i:0, MD:Z:66G6, etc.)
2. データの変換操作とクレンジング
XSの情報を使って、データをクレンジング
ゲノムに複数箇所アライメントされたreadは、どこのゲノム領域を読んだ配列か不確かなので除外する操作。
#クレンジングする前
wc sample.sam
25003
#headerをべつにとっておく
##perlワンライナーを使った方法##
cat sample.sam |perl -e 'while(<>){print $_ if(/\@SQ||\@PG/);}' >sample.header.txt
##samtoolsを使ったやり方の場合###
cat sample.sam | samtools -h >sample.header.txt
#2カ所以上でmappingされるreadの数
grep "XS" sample.sam > sample.multi.sam
wc sample.multi.sam.old
2515
#multi-mappedを除く。
grep -v "XS" sample.sam > sample.sam.old
grep -v "XS" input.sam > input.sam.old
#headerを結合させる
cat sample.header.txt sample.sam.old >sample.uq.sam
cat input.header.txt input.sam.old >input.uq.sam
samからbamファイルへの変換
samファイルのバイナリー形式であるbamファイルへ変換する。
samtools view -bS sample.uq.sam >sample.uq.bam
samtools sort sample.uq.bam sample.uq.sort
samtools view -b -S input.uq.sam >input.uq.bam
samtools sort input.uq.bam input.uq.sort
#ls -alをつかって、ファイルサイズを確認しておく。
ls -al
積極的におすすめ: Picard-toolsを使ったPCR duplicatesの除去
上記までのクレンジングしてもなお、peak callをした時にセントロメアや、テロメア、GCリッチな配列、ホモポリマーなどの領域に大きなpeakが検出されてしまう場合、PCR duplicateの除去を(積極的に)オススメする。
picard-tools MarkDuplicates INPUT=sample.uq.sort.bam OUTPUT=sample.uq.sort.drm.bam METRICS_FILE=sample.uq.sort.metrics AS=true REMOVE_DUPLICATES=true VALIDATION_STRINGENCY=SILENT
picard-tools MarkDuplicates INPUT=input.uq.sort.bam OUTPUT=input.uq.sort.drm.bam METRICS_FILE=input.uq.sort.metrics AS=true REMOVE_DUPLICATES=true VALIDATION_STRINGENCY=SILENT
出力結果
[Mon Aug 03 01:30:21 JST 2015] net.sf.picard.sam.MarkDuplicates INPUT=[sample.uq.sort.bam] OUTPUT=sample.uq.sort.drm.bam METRICS_FILE=sample.uq.sort.metrics REMOVE_DUPLICATES=true ASSUME_SORTED=true VALIDATION_STRINGENCY=SILENT PROGRAM_RECORD_ID=MarkDuplicates PROGRAM_GROUP_NAME=MarkDuplicates MAX_SEQUENCES_FOR_DISK_READ_ENDS_MAP=50000 MAX_FILE_HANDLES_FOR_READ_ENDS_MAP=8000 SORTING_COLLECTION_SIZE_RATIO=0.25 READ_NAME_REGEX=[a-zA-Z0-9]+:[0-9]:([0-9]+):([0-9]+):([0-9]+).* OPTICAL_DUPLICATE_PIXEL_DISTANCE=100 VERBOSITY=INFO QUIET=false COMPRESSION_LEVEL=5 MAX_RECORDS_IN_RAM=500000 CREATE_INDEX=false CREATE_MD5_FILE=false
[Mon Aug 03 01:30:21 JST 2015] Executing as iu@bio on Linux 3.13.0-57-generic amd64; OpenJDK 64-Bit Server VM 1.7.0_79-b14; Picard version: 1.105() JdkDeflater
INFO 2015-08-03 01:30:22 MarkDuplicates Start of doWork freeMemory: 30647024; totalMemory: 31457280; maxMemory: 620756992
INFO 2015-08-03 01:30:22 MarkDuplicates Reading input file and constructing read end information.
INFO 2015-08-03 01:30:22 MarkDuplicates Will retain up to 2463321 data points before spilling to disk.
WARNING 2015-08-03 01:30:22 AbstractDuplicateFindingAlgorithm Default READ_NAME_REGEX '[a-zA-Z0-9]+:[0-9]:([0-9]+):([0-9]+):([0-9]+).*' did not match read name 'SRR445816.33226380'. You may need to specify a READ_NAME_REGEX in order to correctly identify optical duplicates. Note that this message will not be emitted again even if other read names do not match the regex.
INFO 2015-08-03 01:30:25 MarkDuplicates Read 1,000,000 records. Elapsed time: 00:00:02s. Time for last 1,000,000: 2s. Last read position: chr17:53,093,914
INFO 2015-08-03 01:30:25 MarkDuplicates Tracking 0 as yet unmatched pairs. 0 records in RAM.
INFO 2015-08-03 01:30:26 MarkDuplicates Read 1550235 records. 0 pairs never matched.
INFO 2015-08-03 01:30:27 MarkDuplicates After buildSortedReadEndLists freeMemory: 242052288; totalMemory: 365953024; maxMemory: 620756992
INFO 2015-08-03 01:30:27 MarkDuplicates Will retain up to 19398656 duplicate indices before spilling to disk.
INFO 2015-08-03 01:30:27 MarkDuplicates Traversing read pair information and detecting duplicates.
INFO 2015-08-03 01:30:27 MarkDuplicates Traversing fragment information and detecting duplicates.
INFO 2015-08-03 01:30:27 MarkDuplicates Sorting list of duplicate records.
INFO 2015-08-03 01:30:27 MarkDuplicates After generateDuplicateIndexes freeMemory: 347730928; totalMemory: 506462208; maxMemory: 620756992
INFO 2015-08-03 01:30:27 MarkDuplicates Marking 159965 records as duplicates.
INFO 2015-08-03 01:30:27 MarkDuplicates Found 0 optical duplicate clusters.
INFO 2015-08-03 01:30:39 MarkDuplicates Before output close freeMemory: 517976680; totalMemory: 523239424; maxMemory: 620756992
INFO 2015-08-03 01:30:46 MarkDuplicates After output close freeMemory: 507489848; totalMemory: 512753664; maxMemory: 620756992
[Mon Aug 03 01:30:46 JST 2015] net.sf.picard.sam.MarkDuplicates done. Elapsed time: 0.41 minutes.
Runtime.totalMemory()=512753664
積極的にオススメ: blacklistを使ったartifact配列除去
ChIP-seq解析で高頻度にみつかるゲノム領域がblacklistとして公開されている。「InputやIgGコントロールを利用しているから後から除くので安心」などと思わずに、私は先に除くことをお勧めする。経験上そのような部分はInput, IgGコントロールをbackgroundに用いてもpeak callされる。理由は正確に分からないが、塩基バイアスが激しく、とても抗体によるenrichmentとは考えられない領域が多い。おそらくsonicationの不均一biasや、technicalなbiasではないかと考えている。
除去方法はintersectBed等を用いる。
intersectBed -abam sample.uq.sort.bam -b wgEncodeDacMapabilityConsensusExcludable.bed -v > sample.uq.bk.bam
Optional: bamファイルから、ゲノム上のrepeat領域を除く
ChIP-seqデータはゲノムDNAをシェアリングしているので、ホモポリマーをはじめとしたGCリッチな配列や繰り返し配列が検出されやすい。こういった領域の場合、通常のChIP-seq peakのようにはならず、崩れた汚い山になっていることが多い。そこで、reapeatに結合性のタンパク質の場合を除き、reapeat配列にマップされたのリードをあらかじめ除去しておく。 ただし、転写因子によってはrepeat領域に結合性をもつ場合や、promoter領域などにCpGアイランドがある場合に、このfilteringが強烈すぎる場合がある。peakを眺めつつ除去するのが適切か否かは判断する必要がある。これを行う前には、MarkDuplicatesなどのPCR duplicatesを除去しておくと、ある程度このようなartifactは考えなくてよいので、MarkDuplicatesを必ず行った上で判断するとよい。
intersectBed -abam sample.uq.sort.bam -b hg19.rmsk.2.bed -v > sample.uq.rmsk.bam
intersectBed -abam input.uq.sort.bam -b hg19.rmsk.2.bed -v > input.uq.rmsk.bam