ChIP seqデータの可視化 - suimye/NGS_handson2015 GitHub Wiki
#3. アライメント後のデータの可視化
UCSC genome browserは、米国カルフォルニア大学サンタクルズ校が提供するオンラインゲノムブラウザです。ENCODEプロジェクトをはじめ、FANTOM、その他の世界規模のリソースプロジェクトがデータをUCSC上に公開しているので、公共データベースとご自分のデータをweb上で比較をするには便利です。一方で、IGVは、Broad Instituteが提供するゲノムブラウザで、local PCにインストールして利用することができます。tdfファイルとよばれるIGV専用ファイル形式に変換する事で、自分のデータをストレスが少なく閲覧することができる良ツールです。
##3.1. データをUCSC genome browserで見る
genomeCoverageBed -ibam sample.uq.rmsk.bam -bg -trackline -trackopts 'name="sample" color=250,0,0' > sample.bedGraph
genomeCoverageBed -ibam input.uq.rmsk.bam -bg -trackline -trackopts 'name="input" color=250,250,0' > input.bedGraph
#さらに軽快なbigWigファイルへ変換する
bedGraphToBigWig sample.bedGraph /home/genome/hg19/chromInfo.txt sample.bw
bedGraphToBigWig input.bedGraph /home/genome/hg19/chromInfo.txt input.bw
- UCSC genome browserへアクセス
- hg19のゲノムトラックであることを確認
- manageCustomTracksを選択
- add custom tracksでbigWigファイルを選択
- 作成したbedGraphをsubmitする
####Upload中の画面
- uploadが完了すれば次のような画面になるので、go genome browserをクリック
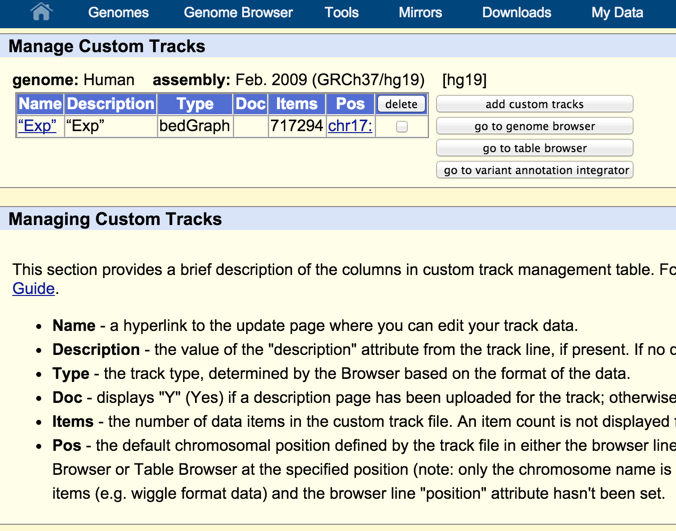
なおこの画面では、自分が入れたファイル(custom track)を管理することができる
- カスタムトラックの追加
- カスタムトラックの削除
- 名前をクリックすれば名前を変更可能
- zoom out x100
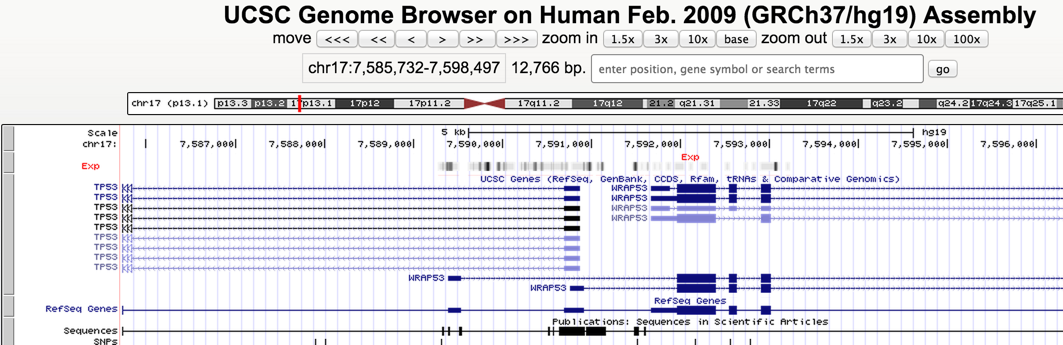
- カスタムトラックのところで、右クリック。denseからfullに変更する
- ブラウザの下にあるCustom Tracksのところからも操作可能
- TP53を入力し、GOを押す。
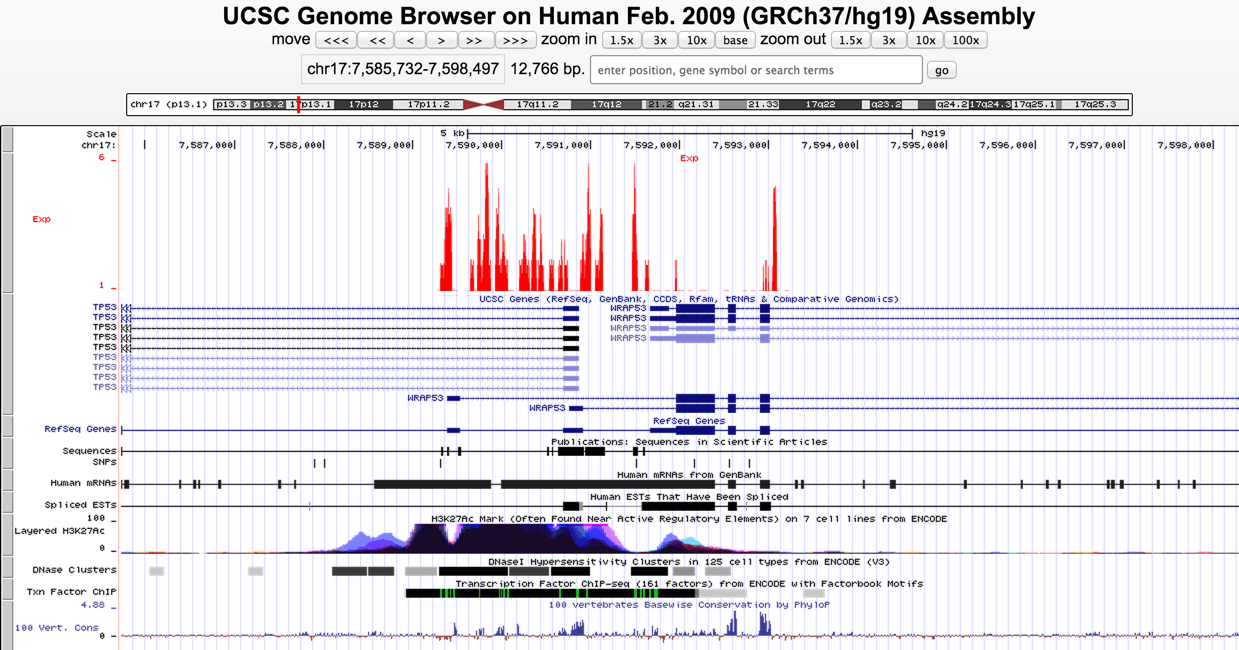
##3.2 IGVの使い方
igvtoolsを用いたtdfファイルへの変換方法について説明する。
準備するもの
- sort済みbamファイル
- IGV genome browserのGUI, もしくは command line tool
コマンドラインツールはこちらからDLして、インストールする(ただしregistrationが必要、現時点(2,2016)でJDK1.7が必要)。
####GUI版、tdfの作り方
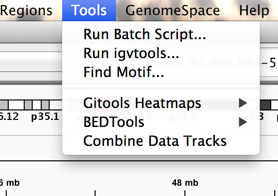
上のように、Tools->Run igvtoolsを選択する。
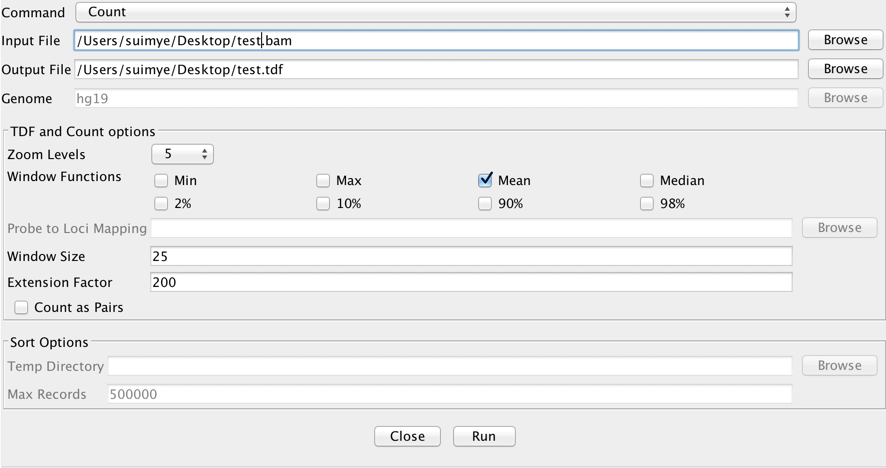
下記のコマンドラインの例と同様のパラメータが設定できるようになっている。window, binサイズなどを指定して、runを押す。
####コマンド例
igvtools count -z 5 -w 25 -e 250 test.sorted.bam test.tdf hg19
引数について
- count: pileup read のcountingでwindow毎の平均値計算を行う。簡単なスライディングウインドでのスコアリングと考えて良い。
- -z: スライディングサイズ。例では5bpごとのoverlapping slideで、window size 25bp内の塩基毎平均タグ数を計算し、その領域のスコアとする。
- -w: windowサイズ。大きいほどへいたんなpeakになるが、スライディングサイズと同様大きいほどファイルサイズは小さくなる。
- -e: tag extend size. そもそものinsert size (sequencingのDNAライブラリ作成時のDNA断片サイズ)を考慮することで、ChIPで沈降したDNAサイズを加味したread sequenceの伸長を行い、peak領域を実際の通りの積み上げになるようにする。sequencingする前に行ったDNA libraryのsize selectionや、最終産物のbioanalyzerの分布などをもとにこのサイズを決めるとよい。
- hg19: ゲノムの種類、バージョンを選択する。igvtoolsを入れたディレクトリ内に/genomes/というフォルダがあるので、その中にあるゲノムであればすぐに利用可能。無い場合は自分で染色体情報を集めて、同ディレクトリに入れておく。
IGVでの可視化の際に、million readで正規化する
表題のままですが、データ間の比較を行うためにmillion readで規格化することが重要です。
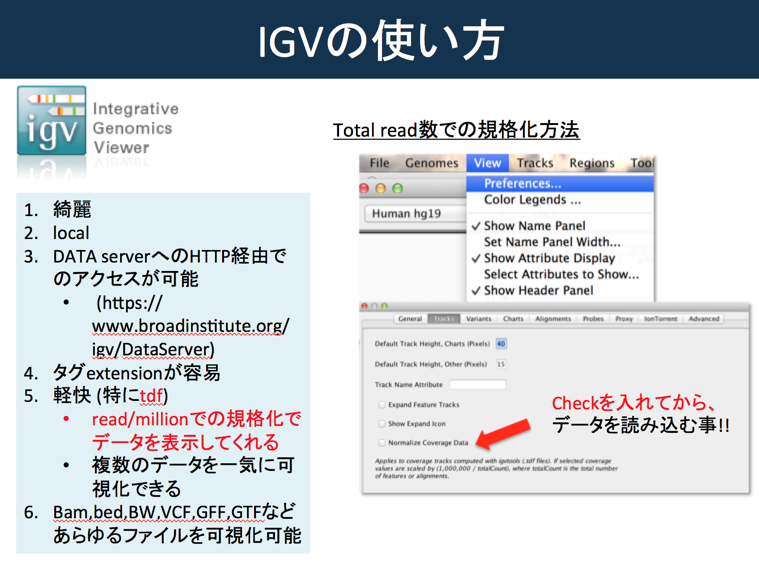
そのためには、tdfファイルにする必要性がありますので (bamファイルでもなるのかな?)、事前にtdfファイルに変換してください。ただし、normalizeしたデータを表示するときは、あらかじめ下記の手順を実施したあとでないと、規格化できていません。
手順
- view
- preferences
- Tracks
- Normalize Coverage Data
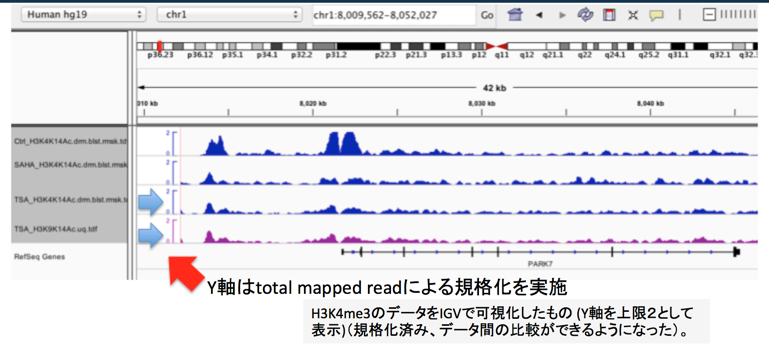
するとこのように、規格化された値で表示されるようになります(比較的に小さな値になってきます)。 試しにpreferenceの変更前後で比較して見るとよいでしょう。