pl1 RNA seq basic - shujishigenobu/omics-collab-cm-nibb GitHub Wiki
この演習ではシロイヌナズナを材料に、RNA-seq解析のパイプラインを学ぶ。シロイヌナズナ(Arabidopsis thaliana)はアブラナ科の植物でモデル植物として広く研究されている。今回の実験では、明暗異なる2条件で生育した植物の遺伝子発現を比較するためにRNA-seq解析を行った。条件は、2D sample (2-day dark;2日間暗環境で生育させた黄色芽生え)と2D2L sample (2-day dark + 2-day light; 2日間暗環境で生育させた後に2日間明環境で生育させた緑化芽生え)の2つ。簡便のため、前者2D2Lサンプルを「明」条件、後者2Dサンプルを「暗」条件と、以下記す。明暗2条件、それぞれ繰り替えし実験を3回行った(biological replicates)。つまり2条件x3反復=6サンプル、となる。それぞれのサンプル植物から定法に従ってRNAを抽出し、ショートリード型次世代シーケンサーIllumina用のRNA-seqライブラリを作成した。完成したライブラリはIllumina社のMiSeqでpaired-end(インサートの両端を読む)の条件で76bpずつシークエンスした。今回の演習で提供するシーケンスデータは、シーケンサーから得られた生リードをすでに前処理 (cutadaptを利用して無駄なアダプター配列や質の低い配列を除去)してある。
この演習の目標は、HISAT2 -> StringTie -> edgeR のパイプラインの学習を通して、明暗条件の差で発現の差がある遺伝子=DEG (differential expressed genes)を同定することである。
このパイプラインは基礎生物学研究所 ゲノムインフォマティクストレーニングコース(GITC)の演習問題 case2 をベースに、作成した。
(参考)
以降の手順はSet up AWSの作業を実施済みであることが前提です。まだ実行環境を準備していない場合はSet up AWSを参考に環境構築を行ってください。
事前に環境構築を準備しインスタンスを停止している場合はインスタンスの起動をしてください。
SSH Remote Connectionを参考に SSH コマンドで仮想マシンへリモート接続を行ってください。
SSH コマンドでリモート接続後、以下のコマンドで演習で必要なソフトウェアをインストール済みの仮想環境へ切り替えしてください。
conda activate tutorial-rnaseq
-
- リードをゲノムにマップする。ソフトウェアは、hisat2を使う。
-
- マッピング結果から遺伝子ごとにリード数をカウントする。ソフトウェアは、stringtieを使う。
-
- 2で得られたカウントデータに基づいて明暗条件の2群間比較の統計解析を行う。ソフトウェアは、edgeRを使う。
データファイルを以下から取得してください。
もし上のURLにうまく接続できない場合は、Google Driveから取得してください。
- pl1-data.tar.gz (261M)
解凍すると、dataフォルダが生成される。dataフォルダ以下に本ハンズオン実習に使うデータファイルが含まれる。dataフォルダごと、使いやすい場所に移動する。以下では、ホームディレクトリ(~/)の直下にdataフォルダを移動してきたものとして解説する。
Input reads
ファイルは、~/data/reads にある。paired-end シーケンスしているので(断片の両方から読んでいる)、6サンプルそれぞれ、2つずつファイルがある(Read1, Read2もしくはforward, reverseと呼ばれる)。
- 2D_rep1: 実験条件=暗, 繰り返し実験#1: 2D_rep1_R1.fastq, 2D_rep1_R2.fastq [R1: Read1 (Fwd); R2: Read2 (Rev) 以下同様]
- 2D_rep2: 実験条件=暗 繰り返し実験#2: 2D_rep2_R1.fastq, 2D_rep2_R2.fastq
- 2D_rep3: 実験条件=暗, 繰り返し実験#3: 2D_rep3_R1.fastq, 2D_rep3_R2.fastq
- 2D2L_rep1: 実験条件=明, 繰り返し実験#1: 2D2L_rep1_R1.fastq, 2D2L_rep1_R2.fastq
- 2D2L_rep2: 実験条件=明, 繰り返し実験#2: 2D2L_rep2_R1.fastq, 2D2L_rep2_R2.fastq
- 2D2L_rep3: 実験条件=明, 繰り返し実験#3, 2D2L_rep3_R1.fastq, 2D2L_rep3_R2.fastq
実験条件正確には、2D=2日間暗、2D2L=2日間暗の後2日間明
Reference
- genome sequence: genome.fa -- シロイヌナズナのゲノムシーケンス。FASTA フォーマット。
- gene annotation: genes.gtf -- シロイヌナズナの遺伝子アノテーション情報。GTF フォーマット。
Software
- hisat2
- stringtie
- samtools
- edgeR
上記環境構築のステップでインストール済み。
作業ディレクトリを作成し、以下の解析はその下で作業しよう。
$ mkdir project-1
$ cd project-1
ファイルにアクセスしやすいようにシンボリックリンクを貼っておこう。
ln -s ../data/genome.fa
ln -s ../data/genes.gtf
ln -s ../data/reads
インストールしたconda環境をactivateする(まだであれば)
conda activate tutorial-rnaseq
解析対象のシーケンスリードの基本情報(リードの本数や長さなど)を取得する。seqkitの stats サブコマンドが便利。
seqkit stats reads/*.fastq.gz
出力結果の例
file format type num_seqs sum_len min_len avg_len max_len
data/reads/2D2L_rep1_R1.fastq.gz FASTQ DNA 539,633 40,955,449 50 75.9 76
data/reads/2D2L_rep1_R2.fastq.gz FASTQ DNA 539,633 40,933,587 50 75.9 76
data/reads/2D2L_rep2_R1.fastq.gz FASTQ DNA 479,469 36,390,170 50 75.9 76
data/reads/2D2L_rep2_R2.fastq.gz FASTQ DNA 479,469 36,371,085 50 75.9 76
data/reads/2D2L_rep3_R1.fastq.gz FASTQ DNA 403,488 30,623,819 50 75.9 76
data/reads/2D2L_rep3_R2.fastq.gz FASTQ DNA 403,488 30,610,016 50 75.9 76
data/reads/2D_rep1_R1.fastq.gz FASTQ DNA 377,791 28,676,854 50 75.9 76
data/reads/2D_rep1_R2.fastq.gz FASTQ DNA 377,791 28,657,121 50 75.9 76
data/reads/2D_rep2_R1.fastq.gz FASTQ DNA 328,491 24,922,717 50 75.9 76
data/reads/2D_rep2_R2.fastq.gz FASTQ DNA 328,491 24,919,185 50 75.9 76
data/reads/2D_rep3_R1.fastq.gz FASTQ DNA 430,418 32,661,169 50 75.9 76
data/reads/2D_rep3_R2.fastq.gz FASTQ DNA 430,418 32,648,214 50 75.9 76
hisat2で検索するためにはリファレンスゲノムの配列をインデックス化する準備が必要
[Ohmura]ファイルパス修正: ./genome.fa(シンボリックリンク) か../data/genome.faのどちらかへ
hisat2-build ../genome.fa genome
genome.1.ht2 など8つのファイルが生成される。
hisat2を使ってリード配列をゲノム(上記で作成したインデックスを利用)にマッピングする。まず、hisat2コマンドの使い方を確認。
hisat2 --help
Usage:
hisat2 [options]* -x <ht2-idx> {-1 <m1> -2 <m2> | -U <r>} [-S <sam>]
<ht2-idx> Index filename prefix (minus trailing .X.ht2).
<m1> Files with #1 mates, paired with files in <m2>.
Could be gzip'ed (extension: .gz) or bzip2'ed (extension: .bz2).
<m2> Files with #2 mates, paired with files in <m1>.
Could be gzip'ed (extension: .gz) or bzip2'ed (extension: .bz2).
<r> Files with unpaired reads.
Could be gzip'ed (extension: .gz) or bzip2'ed (extension: .bz2).
<sam> File for SAM output (default: stdout)
<m1>, <m2>, <r> can be comma-separated lists (no whitespace) and can be
specified many times. E.g. '-U file1.fq,file2.fq -U file3.fq'.
以下略
-x の後に先ほど作成したインデックス名。今回はpaired-endシーケンスなので、-1と-2オプションでそれぞれRead1, Read2のファイル名を指定。出力はsamフォーマットで欲しいので、-Sの後に出力先のファイル名。
(例)
hisat2 -p 4 --dta -x genome -1 reads/2D_rep1_R1.fastq.gz -2 reads/2D_rep1_R2.fastq.gz -S 2D_rep1.sam
その他のオプションの説明
- -p: number of alignment threads。利用している計算機環境に合わせて設定する。今回使っているEC2はvCPUが4つなので4以下を指定。
- --dta: report alignments tailored for transcript assemblers including StringTie. この後、StringTieを使うので、このオプションをつける。
他の5つも同様に。
hisat2 -p 4 --dta -x genome -1 reads/2D_rep2_R1.fastq.gz -2 reads/2D_rep2_R2.fastq.gz -S 2D_rep2.sam
hisat2 -p 4 --dta -x genome -1 reads/2D_rep3_R1.fastq.gz -2 reads/2D_rep3_R2.fastq.gz -S 2D_rep3.sam
hisat2 -p 4 --dta -x genome -1 reads/2D2L_rep1_R1.fastq.gz -2 reads/2D2L_rep1_R2.fastq.gz -S 2D2L_rep1.sam
hisat2 -p 4 --dta -x genome -1 reads/2D2L_rep2_R1.fastq.gz -2 reads/2D2L_rep2_R2.fastq.gz -S 2D2L_rep2.sam
hisat2 -p 4 --dta -x genome -1 reads/2D2L_rep3_R1.fastq.gz -2 reads/2D2L_rep3_R2.fastq.gz -S 2D2L_rep3.sam
hisat2実行時にmappning stats情報がreportされる。これはマッピング率など実験の成否を評価できる数値であるので記録しておくとよい。あるいは以下のように、--new-summary --summary-fileのオプションを付けておくと、mappning stats情報が指定するファイルに出力される。
$ hisat2 -p 4 --dta --new-summary --summary-file 2D_rep1.summary -x genome -1 reads/2D_rep1_R1.fastq.gz -2 reads/2D_rep1_R2.fastq.gz -S 2D_rep1.sam
hisat2の結果ファイルをSAMフォーマットからBAMに変換する。さらにインデックスを作成する。これらの処理には、samtoolsを使う。
samtools sort -@ 3 -o 2D_rep1.sorted.bam 2D_rep1.sam
samtools index 2D_rep1.sorted.bam
他の5つも同様。ちなみに、シェルスクリプトで6つ自動で処理するには以下のようにする。
for f in *sam
do
samtools sort -@ 3 -o `basename $f .sam`.sorted.bam $f
done
for f in *bam
do
samtools index $f
done(編集注:このセクションは改変を検討中。IGVのローカル環境のインストールの説明を上でまだ行っていない。AWSからデータダウンロードには費用がかかる。bam fileの合計サイズを調べると300MBくらい。転送料金5円以下なので許容範囲か。SS)
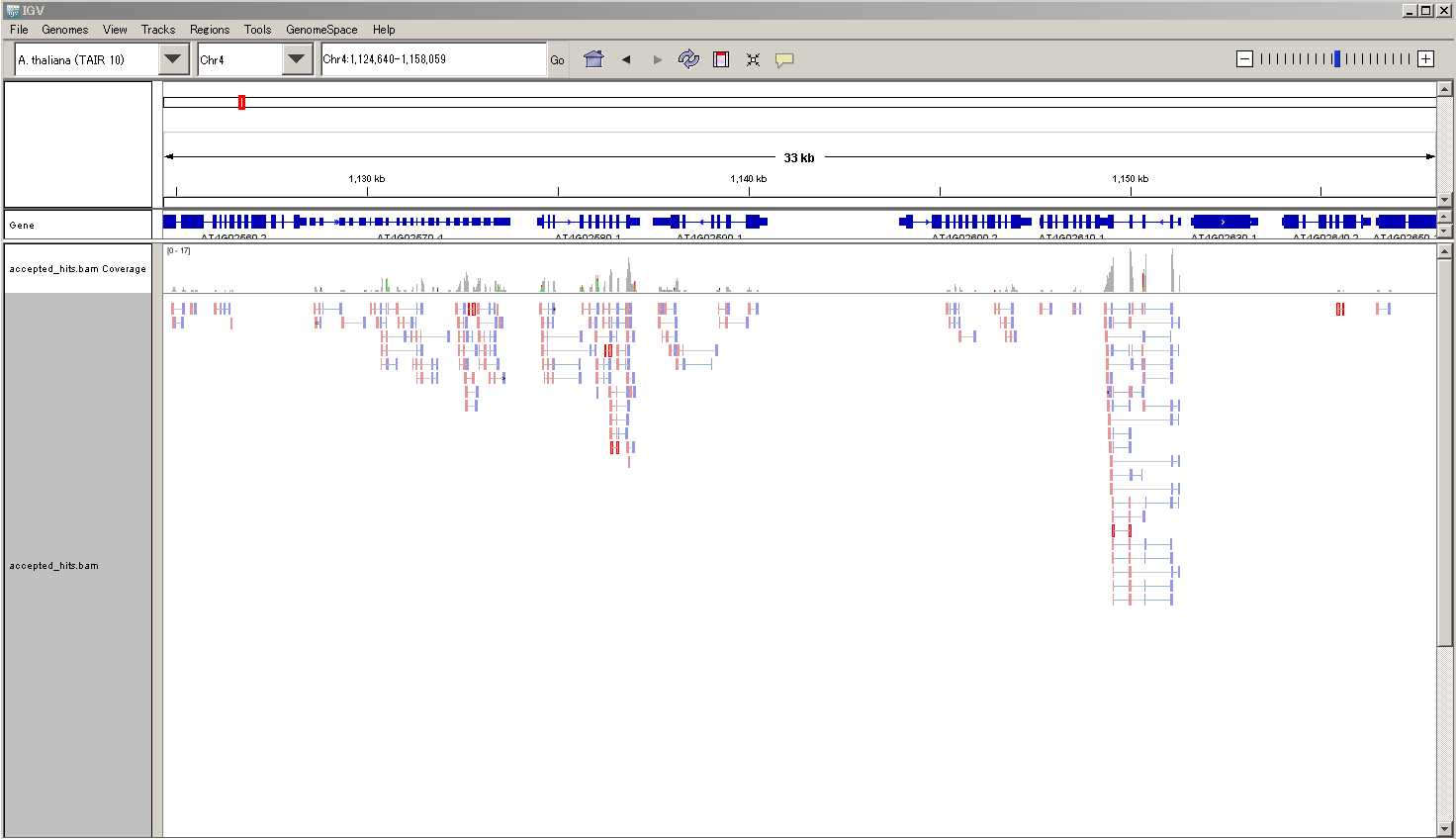
ゲノムブラウザIGV上で、マッピング結果を可視化する。
- 手元のマシンに全ての
sorted.bam及び、sorted.bam.baiファイルをscpコマンドで転送してくること。- 実行コマンド例:
scp -i ~/.ssh/handson.pem ubuntu@[instance-public-dns-name]:/home/ubuntu/project-1/*.bam* .
- 実行コマンド例:
- IGVを立上げる。
- メニュー
Genomes > Load Genome From File...でgenome.faを選択、File > Load from File ...でgenes.gtfを選択 -
File > Load from File ...で作製した.sorted.bamを読み込む - 適当にズームアップする。
stringtie -p 4 -e -G genes.gtf -o stringtie_count/2D_rep1/2D_rep1.gtf 2D_rep1.sorted.bam
stringtie -p 4 -e -G genes.gtf -o stringtie_count/2D_rep2/2D_rep2.gtf 2D_rep2.sorted.bam
stringtie -p 4 -e -G genes.gtf -o stringtie_count/2D_rep3/2D_rep3.gtf 2D_rep3.sorted.bam
stringtie -p 4 -e -G genes.gtf -o stringtie_count/2D2L_rep1/2D2L_rep1.gtf 2D2L_rep1.sorted.bam
stringtie -p 4 -e -G genes.gtf -o stringtie_count/2D2L_rep2/2D2L_rep2.gtf 2D2L_rep2.sorted.bam
stringtie -p 4 -e -G genes.gtf -o stringtie_count/2D2L_rep3/2D2L_rep3.gtf 2D2L_rep3.sorted.bam
-e オプションをつけることにより、-Gで指定したgtfファイルに記載されている遺伝子モデルのみを解析対象にする。(もし-eをつけなければ新規の遺伝子を探索する)。これらのコマンドにより、この後の解析に用いるカウント情報が含まれる新たなgtfファイルが、-oで指定するファイルへ出力される。
上記のstringtieの結果のgtf出力から、遺伝子ごとにリードが何個あるのかのカウントデータをテーブル形式で出力する。つまり,
行:遺伝子数 x 列:条件数
のテーブル(matrix)を作成する。この形式のテーブルは、多くの遺伝子発現解析、発現変動の統計解析やネットワーク解析のツールで入力情報として求められる。
stringtieのgtf出力をカウントデータに変換するには、strinttieの開発者が提供している、prepDE.py スクリプトを利用する。今回はbiocondaセットアップ時にインストール済みであり、パスも通っている。
prepDE.py -i stringtie_count
以下の2つの結果ファイルが生成される。
- gene_count_matrix.csv
- transcript_count_matrix.csv
前者がgene(遺伝子)単位のカウントデータ、後者がtranscript(mRNA、スプライシングバリアンとが存在する場合は別々にカウント)単位のカウントデータ。多くの解析は遺伝子単位で行うので、主に前者のファイルを使う。
less コマンド, wc コマンドで内容確認
(ex)
gene_id,2D2L_rep1,2D2L_rep2,2D2L_rep3,2D_rep1,2D_rep2,2D_rep3
AT1G01020|ARV1,8,8,4,5,0,19
AT1G01060|LHY,3,3,0,0,0,4
AT1G01070|AT1G01070,11,9,4,0,0,0
AT1G01040|DCL1,36,18,22,24,42,18
AT1G01046|MIR838A,0,0,1,0,0,0
AT1G01050|AtPPa1,67,70,54,45,33,25
AT1G01080|AT1G01080,76,58,58,68,38,55
...
wc gene_count_matrix.csv
33603 33680 1140286 gene_count_matrix.csv
カンマ区切りのCSVフォーマットであること、33602 x 6 の matrixであることがわかる。
前ステップまでで得られた遺伝子発現カウントデータに基づいて、2D2L と 2D の2つの条件で統計的に有意に発現レベルが異なる遺伝子を見つける。ここでは、Rのライブラリの一つであるedgeRを用いて2群間比較のパイプラインをR環境で実施する。
コマンドラインにてR環境を起動する。
$ R
以下R環境
> library(edgeR) # edgeRライブラリを読み込む
# 前ステップまでで得られた遺伝子ごとのリードカウントデータ(CSVフォーマット)を読み込む。データフレームに格納される。
> dat <- read.csv("gene_count_matrix.csv",row.names=1)
> head(dat) # 適切に入力できたか確認
X2D2L_rep1 X2D2L_rep2 X2D2L_rep3 X2D_rep1 X2D_rep2 X2D_rep3
MSTRG.1|ARV1 8 8 4 5 0 19
AT1G01060|LHY 3 3 0 0 0 4
AT1G01070|AT1G01070 11 9 4 0 0 0
MSTRG.6|DCL1 36 18 22 24 42 18
MSTRG.6|MIR838A 0 0 1 0 0 0
MSTRG.7|AtPPa1 67 70 54 45 33 25
# ここからedgeRを使った解析
> grp <- c(rep("2D2L", 3), rep("2D", 3)) # 6サンプルがそれぞれ2条件のどちらに属するか定義
> grp
[1] "2D2L" "2D2L" "2D2L" "2D" "2D" "2D"
> D <- DGEList(dat, group=grp) # 先ほど読み込んだカウントデータのデータフレームをedgeRで扱うためのオブジェクトに変換する。
> D <- calcNormFactors(D, method="TMM") # Normalization (標準化)
> D <- estimateCommonDisp(D) # 実データから確率分布(負の二項分布)のパラメータ推定 step-1
> D <- estimateTagwiseDisp(D) # 実データから確率分布(負の二項分布)のパラメータ推定 step-2
# ここまでで準備完了。上記ステップで推定された各種パラメータを確認しておく。
> D$samples # normalization の効果を確認
group lib.size norm.factors
X2D2L_rep1 2D2L 987874 1.0267383
X2D2L_rep2 2D2L 824228 0.9305706
X2D2L_rep3 2D2L 689697 0.9888950
X2D_rep1 2D 597188 1.0250815
X2D_rep2 2D 516354 1.1027216
X2D_rep3 2D 655218 0.9363031
> D$common.dispersion #負の二項分布のcommon dispersion
[1] 0.243539
> summary(D$tagwise.dispersion) #負の二項分布のtagごと(今回の場合場合遺伝子ごとの)dispersion
Min. 1st Qu. Median Mean 3rd Qu. Max.
0.003805 0.218418 0.318444 1.599216 2.219825 9.752049それではいよいよ、発現変動遺伝子(Differential Expressed Genes; DEG)の同定。edgeRのexactTest関数を使う。これは上記のように推定されたデータの確率分布に基づいて、統計的に有意に条件間で発現レベルが異なるかどうかをtestする。
> de.2D.vs.2D2L <- exactTest(D, pair=c("2D", "2D2L"))
# pvalueが上位の遺伝子を見てみる
> topTags(de.2D.vs.2D2L)
Comparison of groups: 2D2L-2D
logFC logCPM PValue FDR
AT2G34430|LHB1B1 10.394710 10.665673 1.995311e-138 6.704643e-134
AT3G01500|CA1 4.743305 11.015099 5.666909e-135 9.520974e-131
AT5G54770|THI1 6.910262 10.734379 2.778929e-131 3.112586e-127
AT2G38530|LTP2 -5.467124 11.523626 1.033709e-127 8.683676e-124
AT4G14130|XTR7 -6.262324 9.393107 5.761200e-126 3.871757e-122
AT5G54190|PORA -12.891697 9.390253 1.750147e-123 9.801404e-120
AT3G21720|ICL -6.505456 11.583401 8.206907e-123 3.939550e-119
AT3G54890|LHCA1 5.230979 11.440747 2.381888e-121 1.000453e-117
AT3G47470|LHCA4 3.859215 11.471514 2.015963e-102 7.526709e-99
AT2G05070|LHCB2 5.400649 9.067715 1.141347e-99 3.835153e-96
# 計算結果をタブ区切りテキストに出力する。
> tmp <- topTags(de.2D.vs.2D2L, n=nrow(de.2D.vs.2D2L))
> write.table(tmp$table, "de.2D.vs.2D2L.txt", sep="\t", quote=F)R環境から抜けて、de.2D.vs.2D2L.txt ファイルが生成されているかを確認。中身をless等で確認。
DEG遺伝子の検証。たとえばTopTagsで第1位の”AT2G34430|LHB1B1”遺伝子に着目しよう。この遺伝子は、リードカウントは以下のようになっている。
(R環境の場合)
> dat["AT2G34430|LHB1B1",]
X2D2L_rep1 X2D2L_rep2 X2D2L_rep3 X2D_rep1 X2D_rep2 X2D_rep3
AT2G34430|LHB1B1 2743 2875 2227 2 2 0(コマンドラインの場合)
$ grep "^AT2G34430|LHB1B1" gene_count_matrix.csv
AT2G34430|LHB1B1,2743,2875,2227,2,2,0
つまり、2D2L条件(明条件)では発現レベルが高く、2D条件(暗条件)でほとんど発現していない。どのような機能の遺伝子なのであろうか?モデル植物であるシロイヌナズナでは遺伝子情報が蓄積されているのでNCBIのデータベースで確認してみる。
- https://www.ncbi.nlm.nih.gov/gene/ -- NCBI Gene データベース
上記データベースの検索ウィンドウに、AT2G34430 のIDを入力してみよう。
「light-harvesting chlorophyll-protein complex II subunit B1」とアノテーションされており、説明を読むと、葉緑体内部のクロロフィルを構成する光合成に関わるタンパク質をコードする遺伝子であることがわかる。明条件でのみ発現が高かったことが納得いく結果である。
hisat2
https://ccb.jhu.edu/software/hisat2/index.shtml
stringtie
http://www.ccb.jhu.edu/software/stringtie/
edgeR
http://bioconductor.org/packages/release/bioc/html/edgeR.html
論文情報
Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown https://www.nature.com/articles/nprot.2016.095