NextStrain solo con muestras de Peru - quipupe/from-assembly-to-nextstrain GitHub Wiki
Para obtener tanto las secuencias de este tutorial como los metadatos, descargar los archivos de este link https://drive.google.com/drive/folders/1J5uqcreJo3vgPB4X-fZOhZAGH3kXI2od?usp=sharing
Otra forma de descargar los datos actualizados es usar la base de datos GISAID. https://www.epicov.org/epi3/ Necesitarás crear un usuario la primera vez. También descarga la metadata en formato tsv
Las secuencias originales tienen este formato >hCoV-19/Peru/010/2020|EPI_ISL_415787|2020-03-10 y lo preferible sería cambiarlo a un nombre más sencillo para ello usaré expresiones regulares en Notepad++ \|.* borrará todo después de | y luego eliminar hCoV-19
A la metadata es mejor ponerle los nombres de las variables en minúsculas
strain virus accession date location country city host gender age status passage specimen lineage clade
estos cambios ya fueron realizados en el archivo metadata.tsv

Copiar los archivos metadata.tsv y sequences.fasta a una carpeta nueva, llamada ```datadesde donde se correráauspice``.
Filtrar las secuencias
augur filter \
--sequences data/sequences.fasta \
--metadata data/metadata.tsv \
--output results/filtered.fasta \
--group-by city year month \
--min-date 2019
0 sequences were dropped during filtering
0 of these were dropped because of their date (or lack of date)
34 sequences have been written out to results/filtered.fasta
Otros metadatos
colors.tsv #Tratar de usar un esquema colorblind
city lima #CC79A7
city huanuco #D55E00
city ica #0072B2
city arequipa #009E73
city lambayeque #E69F00
lat_longs.tsv #En grados decimales
city Lima -12.04318 -77.02824
city Ica -14.06777 -75.72861
city Huanuco -9.93062 -76.24223
city Arequipa -16.39889 -71.535
city Lambayeque -6.70111 -79.90611
Alinear las secuencias
augur align \
--sequences results/filtered.fasta \
--reference-sequence config/coronavirus.gb \
--output results/aligned.fasta \
--fill-gaps\
--nthreads 6\
#Este último comando controla los procesadores, solo está activo para ciertos comandos.
using mafft to align via:
mafft --reorder --anysymbol --nomemsave --adjustdirection --thread 1 results/aligned.fasta.to_align.fasta 1> results/aligned.fasta 2>
results/aligned.fasta.log
Katoh et al, Nucleic Acid Research, vol 30, issue 14
https://doi.org/10.1093%2Fnar%2Fgkf436
No gaps in alignment to trim (with respect to the reference, NC_045512.2)
Construir el árbol
augur tree \
--alignment results/aligned.fasta \
--output results/tree_raw.nwk \
----nthreads 6\
Building a tree via:
iqtree -ninit 2 -n 2 -me 0.05 -nt 1 -s results/aligned-delim.fasta -m GTR > results/aligned-delim.iqtree.log
Nguyen et al: IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies.
Mol. Biol. Evol., 32:268-274. https://doi.org/10.1093/molbev/msu300
Building original tree took 0.15984201431274414 seconds
Calibrar el árbol
augur refine \
--tree results/tree_raw.nwk \
--alignment results/aligned.fasta \
--metadata data/metadata.tsv \
--output-tree results/tree.nwk \
--output-node-data results/branch_lengths.json \
--timetree \
--coalescent opt \
--date-confidence \
--date-inference marginal \
--clock-filter-iqd 4
augur refine is using TreeTime version 0.7.6
/home/pipo/.local/lib/python3.8/site-packages/treetime/aa_models.py:88: VisibleDeprecationWarning: Creating an ndarray from ragged nested
sequences (which is a list-or-tuple of lists-or-tuples-or ndarrays with different lengths or shapes) is deprecated. If you meant to do this, you
must specify 'dtype=object' when creating the ndarray
_BLOSUM45 = np.array([
0.87 TreeTime.reroot: with method or node: least-squares
0.87 TreeTime.reroot: rerooting will ignore covariance and shared ancestry.
0.92 TreeTime.reroot: with method or node: least-squares
0.92 TreeTime.reroot: rerooting will ignore covariance and shared ancestry.
pruning leaf NC_045512.2
0.98 WARNING: Previous versions of TreeTime (<0.7.0) RECONSTRUCTED sequences of
tips at positions with AMBIGUOUS bases. This resulted in unexpected
behavior is some cases and is no longer done by default. If you want to
replace those ambiguous sites with their most likely state, rerun with
`reconstruct_tip_states=True` or `--reconstruct-tip-states`.
1.14 TreeTime.reroot: with method or node: least-squares
1.14 TreeTime.reroot: rerooting will account for covariance and shared ancestry.
1.27 ###TreeTime.run: INITIAL ROUND
2.22 TreeTime.reroot: with method or node: least-squares
2.22 TreeTime.reroot: rerooting will account for covariance and shared ancestry.
2.28 ###TreeTime.run: rerunning timetree after rerooting
3.25 ###TreeTime.run: ITERATION 1 out of 2 iterations
5.50 ###TreeTime.run: ITERATION 2 out of 2 iterations
14.16 ###TreeTime.run: FINAL ROUND - confidence estimation via marginal
reconstruction
Inferred a time resolved phylogeny using TreeTime:
Sagulenko et al. TreeTime: Maximum-likelihood phylodynamic analysis
Virus Evolution, vol 4, https://academic.oup.com/ve/article/4/1/vex042/4794731
updated tree written to results/tree.nwk
node attributes written to results/branch_lengths.json
Reconstruir características ancestrales
augur traits \
--tree results/tree.nwk \
--metadata data/metadata.tsv \
--output results/traits.json \
--columns city
augur traits is using TreeTime version 0.7.6
Assigned discrete traits to 34 out of 34 taxa.
NOTE: previous versions (<0.7.0) of this command made a 'short-branch
length assumption. TreeTime now optimizes the overall rate numerically
and thus allows for long branches along which multiple changes
accumulated. This is expected to affect estimates of the overall rate
while leaving the relative rates mostly unchanged.
Inferred ancestral states of discrete character using TreeTime:
Sagulenko et al. TreeTime: Maximum-likelihood phylodynamic analysis
Virus Evolution, vol 4, https://academic.oup.com/ve/article/4/1/vex042/4794731
results written to results/traits.json
Reconstruir secuencias ancestrales
augur ancestral \
--tree results/tree.nwk \
--alignment results/aligned.fasta \
--output-node-data results/nt_muts.json \
--inference joint
augur ancestral is using TreeTime version 0.7.6
/home/pipo/.local/lib/python3.8/site-packages/treetime/aa_models.py:88: VisibleDeprecationWarning: Creating an ndarray from ragged nested
sequences (which is a list-or-tuple of lists-or-tuples-or ndarrays with different lengths or shapes) is deprecated. If you meant to do this, you must specify 'dtype=object' when creating the ndarray
_BLOSUM45 = np.array([
Inferred ancestral sequence states using TreeTime:
Sagulenko et al. TreeTime: Maximum-likelihood phylodynamic analysis
Virus Evolution, vol 4, https://academic.oup.com/ve/article/4/1/vex042/4794731
ancestral mutations written to results/nt_muts.json
Traducir las secuencias a amino ácidos
augur translate \
--tree results/tree.nwk \
--ancestral-sequences results/nt_muts.json \
--reference-sequence config/coronavirus.gb \
--output results/aa_muts.json
Read in 12 features from reference sequence file
amino acid mutations written to results/aa_muts.json
Exportar con Augur
augur export v2 \
--tree results/tree.nwk \
--metadata data/metadata.tsv \
--node-data results/branch_lengths.json \
results/traits.json \
results/nt_muts.json \
results/aa_muts.json \
--colors config/colors.tsv \
--lat-longs config/lat_longs.tsv \
--auspice-config config/auspice_config.json \
--output auspice/coronavirus.json
Validating schema of 'results/aa_muts.json'...
Validating config file config/auspice_config.json against the JSON schema
Validating schema of 'config/auspice_config.json'...
WARNING: These values for trait city were not specified in your provided color scale: lima, arequipa, huanuco, ica. Auspice will create colors for them.
Validating produced JSON Validating schema of 'auspice/coronavirus.json'... Validating that the JSON is internally consistent...
Validation of 'auspice/coronavirus.json' succeeded.
Para resolver el problema de los colores chequear colors.tsv y darle el formato correcto con tabs
city lima #56B4E9
city huanuco #E69F00
city ica #CC79A7
city arequipa #E68234
city lambayeque #E1512A
Validating schema of 'results/aa_muts.json'...
Validating config file config/auspice_config.json against the JSON schema
Validating schema of 'config/auspice_config.json'...
Validating produced JSON
Validating schema of 'auspice/coronavirus.json'...
Validating that the JSON is internally consistent...
Validation of 'auspice/coronavirus.json' succeeded.
Observar en Ausipice
auspice.js view --datasetDir auspice/
luego entrar a internet a esta dirección
http://localhost:4000/coronavirus
Otros datos
Tuve que eliminar la etiqueta "/locus_tag" del archivo .gb /locus_tag="\w+" porque no nombraba bien los genes.
Es mejor usar como genoma de referencia https://www.ncbi.nlm.nih.gov/nuccore/MN908947 la secuencia de GenBank y no la de RefSeq
Auspice o como ser vería en NextStrain
Arbol con escala en días

También es importante ver el árbol pero con escala en distancia genética (divergence). Por ejemplo los casos 01-2, 002, 003, 004, 009, 013 son la misma secuencia. Esos casos son del vuelo de LAN, apuesto un pasaje a Iquitos!

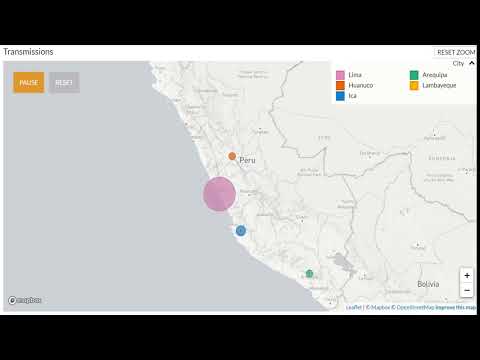
El mapa muestra como se relacionan estas secuencias, se puede dar play y ver como ha ido avanzando la epidemia. Debemos hacer un GIF de esto.

Las variantes genéticas se mapean sobre el genoma de referencia

Finalmente, se pueden establecer filtros, esto depende de la metadata. Ver el archivo .json en la carpeta compartida
https://drive.google.com/drive/folders/18dwnAxU-wncQJj1rGQCkdHAmLj0WeD9Q?usp=sharing
Ruta de transmisión (Video, dar click en el link o en la imagen) https://youtu.be/BqhgdytQdlQ