Tutorial 1 - mjsull/HapFlow GitHub Wiki
Real data from mixed-strain infection of Koala.
This tutorial will let you explore a premade flow file and show you how to separate reads into two operational taxonomic units. In the interest of keeping the example files small only reads aligned to bases 360,000..380,000 were used. This tutorial assumes you have downloaded and extracted tutorial1.zip from the HapFlow website.
- Launch HapFlow by double clicking on the .exe or app file.
- Load flow by selecting File > Load flows and then choosing example1.flw
- Explore the flow file by scrolling from left to right.
- Hide lines marking flows with gaps by selecting view > hide gapped.
- Adjust the amount of variants displayed by pressing A and D on your keyboard.
- Adjust the height of the flows by pressing W and S on your keyboard.
- Left click on a Flow to highlight the flow, right click to bring up options relating to the flow.
- Go to the variant at base 363,350 by selecting tools > goto base and typing in 363350
OSX and GNU/Linux only
- Define OTU 1 by double clicking variants along the top and middle rows.
- Define OTU 2 by selecting tools > Select OTU > 2 and then double clicking variants along the top and middle rows.
- Define OTU 3 by selecting tools > Select OTU > 3 and then double clicking positions of the flow with an erroneous base.
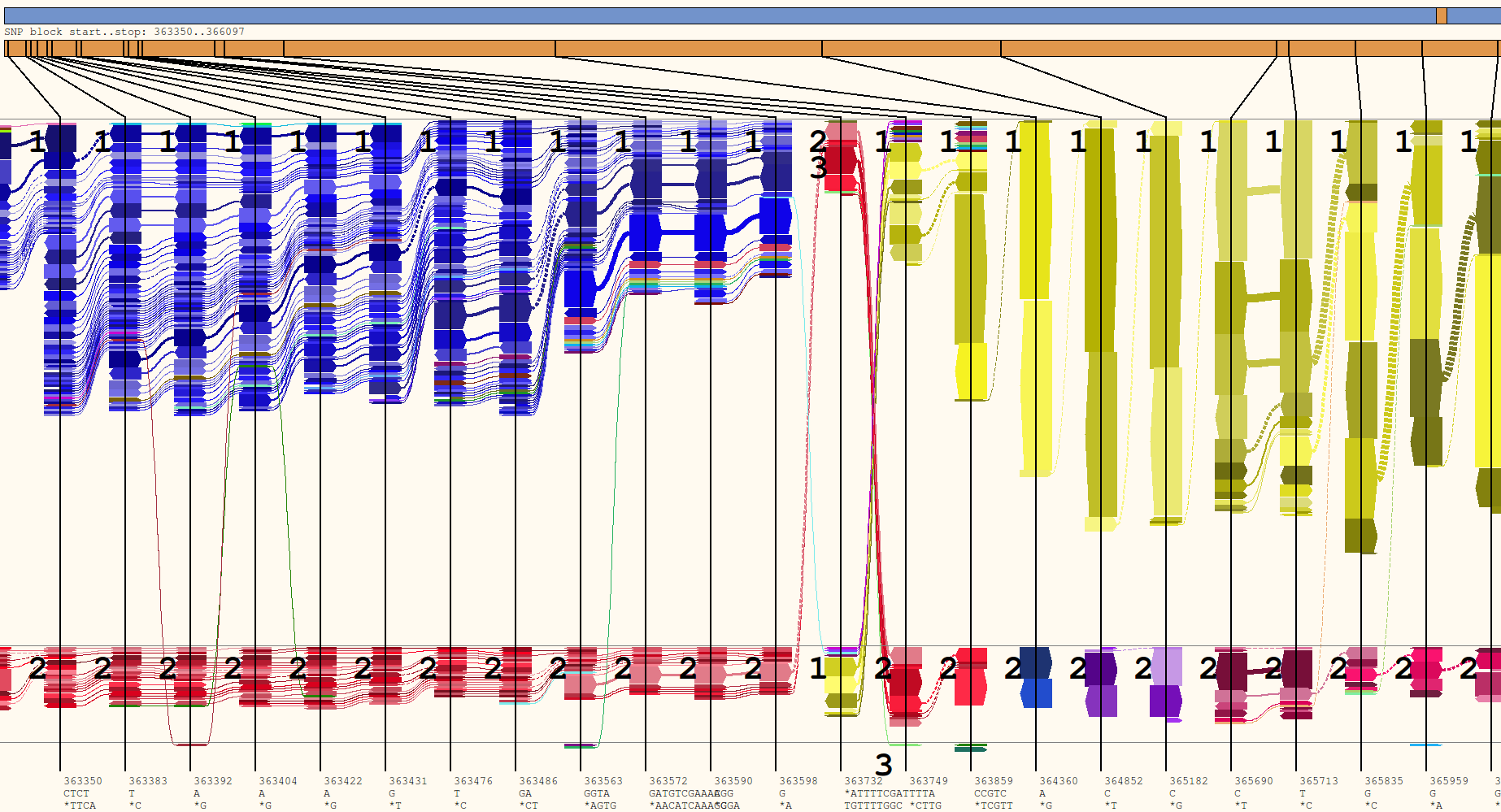 Chlamydia sample with selected OTUs
Chlamydia sample with selected OTUs
- Write OTUs to BAM by selecting tools > Write OTUs and then choosing the downloaded BAM file and defining a prefix for output file. Files will be written to prefix.1.bam prefix.2.bam and prefix.3.bam