TranscriptomeAsmPB - aechchiki/SIB_LongReadsWorkshop_Zurich18 GitHub Wiki
PacBio: IsoSeq
Section: Long-reads Transcriptome [2/4].
The Iso-Seq pipeline is a method to get a high-confidence transcriptome from PacBio RNA-seq data. Here, we will just give you a flavor on how data from Iso-Seq look like, and describe the workflow we went through to get the data you'll be working with.
If you ever need a full tutorial, here is a really good one - directly from PacBio.
Get a high confidence transcripts set
To get the high-confidence transcripts set, we started with an experimental pipeline with size selection (BluePippin Size Selection System), resulting in libraries for three size fractions (1-2kb, 2-3kb and 3-7kb). The Iso-Seq pipeline was run independently on the three size fractions, using merged raw reads from the two SMRTcells per fraction as input.
Size selection is recommended to get the best out from your libraries: it allows to detect a broader and accurate range of transcripts, but also leads to a depletion in very short (for the shortest fraction) and very long transcripts (for the longest fraction). So consider this before starting your library preparation.
We then run the Iso-Seq pipeline on the three fractions, separately, and then merged the final files to get the final isoforms set. This behaviour could reduce the number of full-length isoforms in output. However, we chose this approach since it is advised to obtain a better polished consensus at the end. Two potential pitfalls of this approach are that, (1) the quality of the consensus obtained by ICE itself could be reduced, and (2) the runtime of the whole Iso-Seq pipeline could be longer.
But, what's the Iso-Seq pipeline?
The Iso-Seq pipeline is a set of software developed by PacBio that allows to go from raw reads (in bax.h5/bas.h5 format) and generate the high-confidence transcriptome.
Here's an overview of it:
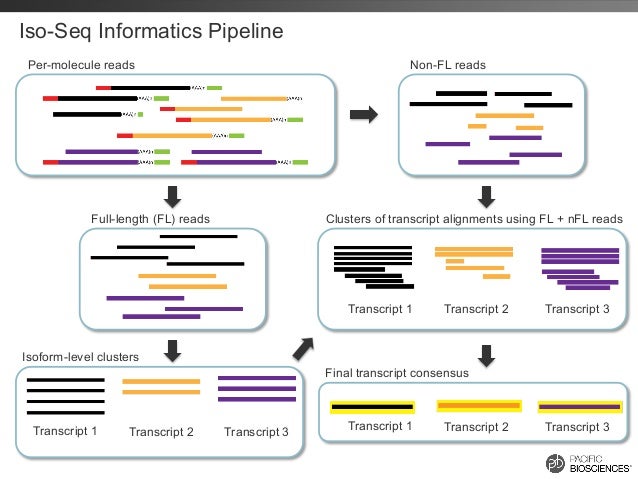
- In the first step, subreads are assembled generating Reads Of Insert (ROI), the single highest quality consensus per insert, and saved to fastq format.
- In the second step ("Classify"), the ROI were determined as full-length non-chimeric (FL) or non full-length (nFL), depending on the presence or not of both 5' and 3' primers at the read ends.
- In the third step ("Cluster"), the FL ROIs were fed into ICE (Iterative Clustering and Error correction) to generate clusters of isoforms, while nFL reads were attributed to the clusters a posteriori.
- In the fourth step, the clusters were polished using Quiver: the final transcript isoform consensus was generated, and saved in fastq format.
What is the output of the Iso-Seq pipeline?
And here's an overview of the structure you get from a real run:
aechchik@frt:/scratch/beegfs/monthly/aechchik/isoforms/cDNA_1-2k_Isoseq$ find . *
.
./qc_report.pdf
./Classify
./Classify/aligned_flnc_reads_of_insert.bam
./Classify/isoseq_primer_info.csv
./Classify/classify_summary.txt
./Classify/isoseq_nfl.fasta
./Classify/isoseq_flnc.fasta
./Classify/isoseq_draft.fasta
./Cluster
./Cluster/cluster_summary.txt
./Cluster/polished_low_qv_consensus_isoforms.fastq
./Cluster/polished_high_qv_consensus_isoforms.fastq
./Cluster/isoseq_cluster_info.csv
./Cluster/consensus_isoforms.fasta
./Cluster/aligned_consensus_isoforms.bam
./reads_of_insert.fastq
Can you tell from which IsoSeq stage did those files come from? (the picture above is here to help)
So, let's take a look of how a set of high-quality isoforms from PacBio looks like. We generated for you a subset of the file we got from Iso-Seq data (corresponding to the polished_high_qv_consensus_isoforms.fastq):
wget https://drive.switch.ch/index.php/s/fhH7oLIQ95EGuPt/download -O pacbioIS_subset.tar.gz
Well, yes. Actually that's it!
At this stage, we can't do better than consider as "transcriptome" the content of the ./Cluster/polished_high_qv_consensus_isoforms.fastq file (the one you just downloaded). Many methods are being developed alongside this main PacBio product, but they are mostly niche-specific (metagenomics, single-gene transcripts...).
This is still the best (and unique) way to get the best transcriptome-wide long-read dataset so far!