Technologies - aechchiki/SIB_LongReadsWorkshop_Zurich18 GitHub Wiki
Section: Introduction [1/3].
We will discuss here PacBio and Oxford Nanopore sequencing technology, in a concise manner, with enough detail to deal with data later on.
Two main sequencing platforms are currently commercialized by PacBio: RSII and Sequel. The data provided by those platforms changes in the format of delivery (which we will explore later on), but not in the sequencing principle.
PacBio sequencing is based on the SMRT (Single Molecule Real Time) sequencing. Here you can find a video briefly explaining it.
In short, SMRT sequencing happens in microwells called ZMWs (Zero Mode Waveguides) directly on the sequencing flowcell (SMRTcell). At the bottom of each ZMWs, a natural polymerase incorporates complementary bases to a DNA fragment. The polymerase continuously 'reads' the fragment to be sequenced, which has to be prepared with hairpin adapters (SMRTbell), where the polymerase can bind to start reading the template. SMRTbells are ligated on both sides of each DNA fragment (double-stranded), forming a circular sequence.
The complete sequence is called a "polymerase read". After the adapter sequences are removed, the sequence is split into "subreads". The consensus of subreads is called a "circular consensus read" (CCS) or "read of insert" (ROI).
Note The fragments to be sequenced can differ in length. For a given sequencing time, short DNA fragments will be read many times, while long fragments may be read just few times completely. This will become particularly important when dealing with transcriptome data (cDNA).
Here is a visual overview of the PacBio reads:

(updated October 2018)
RSII:
- generates up to 1Gbp per SMRTcell, each with 150'000 ZMWs
- average read length: half of reads longer than 20k and some reads longer than 60k
- some hours workflow
Sequel:
- generates up to 20 Gb per SMRTcell (average 8-10Gbp), each with 1'000'000 ZMWs
- average read lengths: up to 30 kb
- <1 day workflow
Both:
- are very big expensive machines (aboslute need for a sequencing facility)
- achieve high consensus accuracies (>99.999%) for whole genome sequencing high fidelity (>99% accuracy) for amplicon and RNA sequencing
- provide data free of systematic errors
There are many ONT platforms available these days (as it was introduced in the morning talks), each with its own applications. The data provided by those platforms is pretty much of the same type.
Here you can find a video, briefly explaining the technology.
Sequencing calls on a MinION platform are based on the detection of electric signal recorded through the nanopores of the flowcell, as the DNA/cDNA fragment passes through it. The nanopores are distributed all over the surface of the MinION flowcell. The fragments to be sequenced are processed with library preparation, then added on the flowcell, and they are guided inside the nanopores by an enzyme standing on top of them. These enzymes unwind the fragments (double-stranded) at the aperture of the nanopore, so a single-stranded DNA can pass through it. While passing through the nanopore, the electrical current is disrupted by the charges of the nucleotides that pass through it. The disruption of charge defines then the nature of the nucleotides flowing through the nanopore.
Depending on the type of nanopores on the flowcell, also called "chemistry", you might encounter different "consensus" data type (mainly 2D and 1D²). The sequencing principle, though, remains the same. In this tutorial, we will deal with data provided by MinION sequencer.
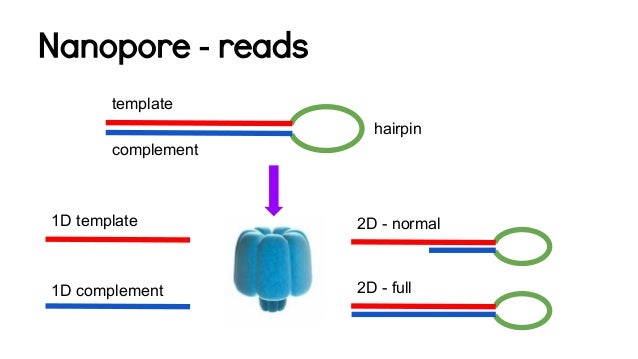
Here is an overview of the ONT reads:
{kind=link}
After DNA fragmentation, the MinION library is prepared by ligating adapters to the double-stranded DNA fragments. One side receives a Y-form adapter, and the other side a hairpin-form adaptor. When the Y-form adapter approaches the pore, one strand starts to be sequenced. This sequence is called "template". After the hairpin-form adapter is sequenced, the "complement" sequence is read. The template and complement sequences are called 1D reads. The of template and complement sequences is called a 2D read.
The 1D² method is basically 1D read with 2Daccuracy. A strand of DNA is sequenced, and always followed by the complementary strand (while in the 2D consensus, some of the reads could not have their complement sequenced, lowering the accuracy). Here is a video explaining the process.
(updated October 2018)
MinION:
- personal sequencer, can be plugged in a laptop
- runtime 1 min - 48 hrs
- up to 30 Gbp yield, and up to 512 channels for simultaneous sequencing
- increased from < 60% to > 85% (over the last 2 years)