04_vasp能带计算 - Yiwei666/13_vasp GitHub Wiki
- 计算流程
- Materials project获取POSCAR、INCAR等文件
- 按照POSCAR中元素顺序生成POTCAR文件
- 静态自洽计算获取CHGCAR文件
- vaspkit导出高对称性 KPOINTS 和 基于 Primitive Cell 的 POSCAR 文件
- 修改INCAR文件,vasp进行非自洽计算(NSCF)能带任务
- vaspkit导出BAND.dat、KLABELS和BAND_GAP等文件
- origin绘制能带
- 注意事项
-
能带计算的KPOINTS与普通计算的KPOINTS不一样,通常需要第一布里渊区内的一条或几条高对称点路径来计算能带性质。传统的做法是通过SeeK-Path网站或者Material Studio软件获得晶体倒易空间第一布里渊区内的高对称点,再通过脚本插值生成高对称点路径上的K点,得到满足要求的KPOINTS。
-
VASP能带计算需要进行自洽计算和非自洽计算两个步骤。在自洽计算中,VASP通过迭代求解波函数和电荷密度的变化来达到自洽,从而得到体系的基态能量和波函数。自洽计算结束后,VASP会输出CHGCAR文件,其中包含了体系的电荷密度信息。在非自洽计算中,VASP会读取上一步的CHGCAR文件,并根据用户指定的参数进行计算。能带计算是一种非自洽计算,需要读入上一步的CHGCAR文件。因此,基于自洽计算得到的CHGCAR文件是进行VASP能带计算的必要条件。在进行VASP能带计算之前,需要先进行自洽计算,得到基态能量和波函数。只有在基态能量和波函数已知的情况下,才能进行能带计算。
- 输入文件
静态自洽计算通常需要以下输入文件
INCAR # 计算的方式和参数
KPOINTS # 定义了 Brillouin 区域内的 K 点网格,用于对电子结构进行采样
POSCAR # 包含了晶体结构的信息,包括原子种类、坐标、晶胞信息等
POTCAR # 包含了与体系中每个元素相关的赝势
sub.sh # 超算提交脚本
- 静态自恰计算输出文件
.
├── AECCAR0
├── AECCAR1
├── AECCAR2
├── CHG
├── CHGCAR
├── CONTCAR
├── DOSCAR
├── EIGENVAL
├── IBZKPT
├── INCAR
├── KPOINTS
├── LOCPOT
├── OSZICAR
├── OUTCAR
├── PCDAT
├── POSCAR
├── POTCAR
├── PROCAR
├── REPORT
├── slurm-2675593.out
├── sub.sh
├── vasprun.xml
├── WAVECAR
└── XDATCAR
- 为什么使用vasp进行能带计算之前需要先静态自洽计算,获得CHGCAR文件?
在进行能带计算之前进行静态自洽计算并获得CHGCAR文件的原因主要是为了提供初始的电荷密度分布。
在静态自洽计算中,VASP通过迭代求解Kohn-Sham方程来找到系统的基态电荷密度。这个基态电荷密度将用作后续计算的初始猜测。在能带计算中,电子的运动状态是通过解Kohn-Sham方程得到的,而这个方程的解依赖于初始的电荷密度。
CHGCAR文件包含了系统的电荷密度信息,它记录了每个原子周围的电荷分布。在进行能带计算时,这个文件用于提供初始的电荷密度分布,以便在新的k点网格上重新解Kohn-Sham方程。这有助于加速能带计算的收敛过程,因为它提供了一个更好的初始猜测。
总体而言,通过在静态自洽计算中获得CHGCAR文件,能够为后续的能带计算提供一个更准确和有效的起点。
- 为什么能带计算比自洽计算要求的K点数要高一些?
在VASP计算中,能带计算比自洽计算要求的K点数要高一些的原因是为了更准确地描述电子能带结构。自洽计算主要用于确定体系的基态电子结构,而能带计算则是在自洽计算的基础上,通过对一系列K点进行计算,得到体系的能带结构。
在自洽计算中,只需要对少量的K点进行计算即可得到准确的电子结构信息。然而,在能带计算中,为了更全面地描述电子能带结构,需要对更多的K点进行计算。通过增加K点数目,可以更好地揭示体系中电子的行为和性质。
- 自洽计算和非自洽计算的区别是什么?为什么能带计算不需要自洽计算?
自洽计算和非自洽计算的区别主要在于是否考虑了电子与电子之间的相互作用。自洽计算考虑了电子与电子之间的相互作用,通过迭代求解波函数和电荷密度的变化来达到自洽,从而得到体系的基态能量和波函数。非自洽计算则不考虑电子与电子之间的相互作用,而是直接使用已知的波函数或电荷密度进行计算。
能带计算不需要自洽计算的原因是,能带计算只需要计算电子在晶格中的能量,而不需要计算电子与电子之间的相互作用。在自洽计算中,电子与电子之间的相互作用会影响波函数的形状,从而影响能带的计算。而非自洽计算则可以忽略电子与电子之间的相互作用,从而直接计算能带。
在VASP中,能带计算是一种非自洽计算。在进行能带计算之前,需要先进行自洽计算,得到基态能量和波函数。只有在基态能量和波函数已知的情况下,才能进行能带计算。
- V2PC静态自洽计算INCAR文件
ALGO = Normal
EDIFF = 1e-05
ENCUT = 520
IBRION = -1
ISIF = 3
ISMEAR = -5
ISPIN = 2
KPAR = 8
LAECHG = True
LASPH = True
LCHARG = True
LORBIT = 11
LREAL = Auto
LVHAR = True
LWAVE = False
MAGMOM = 4*0.003 4*-0.0
NELM = 100
NPAR = 4
NSW = 0
PREC = Accurate
SIGMA = 0.05
SYSTEM = Mp-8044
- V2PC静态自洽计算KPOINTS文件
Automatic kpoint scheme
0
Gamma
9 9 2
- 操作步骤
在含有POSCAR文件的目录下执行以下命令
vaspkit --> 3 --> 303
- vaspkit生成文件目录
.
├── HIGH_SYMMETRY_POINTS
├── INCAR
├── KPATH.in
├── POSCAR
├── POTCAR
├── PRIMCELL.vasp
└── SYMMETRY
- KPATH.in文件内容
K-Path Generated by VASPKIT.
20
Line-Mode
Reciprocal
0.0000000000 0.0000000000 0.0000000000 GAMMA
0.5000000000 0.0000000000 0.0000000000 M
0.5000000000 0.0000000000 0.0000000000 M
0.3333333333 0.3333333333 0.0000000000 K
0.3333333333 0.3333333333 0.0000000000 K
0.0000000000 0.0000000000 0.0000000000 GAMMA
0.0000000000 0.0000000000 0.0000000000 GAMMA
0.0000000000 0.0000000000 0.5000000000 A
0.0000000000 0.0000000000 0.5000000000 A
0.5000000000 0.0000000000 0.5000000000 L
0.5000000000 0.0000000000 0.5000000000 L
0.3333333333 0.3333333333 0.5000000000 H
0.3333333333 0.3333333333 0.5000000000 H
0.0000000000 0.0000000000 0.5000000000 A
0.5000000000 0.0000000000 0.5000000000 L
0.5000000000 0.0000000000 0.0000000000 M
0.3333333333 0.3333333333 0.5000000000 H
0.3333333333 0.3333333333 0.0000000000 K
- 终端显示
===================== Structural Utilities ======================
1) VASP Input-Files Generator 2) Mechanical Properties
3) K-Path for Band-Structure 4) Structure Editor
5) Catalysis-ElectroChem Kit 6) Symmetry Analysis
7) Materials Databases 8) Advanced Structure Models
===================== Electronic Utilities ======================
11) Density-of-States 21) Band-Structure
23) 3D Band-Structure 25) Hybrid-DFT Band-Structure
26) Fermi-Surface 28) Band-Structure Unfolding
31) Charge-Density Analysis 42) Potential Analysis
51) Wave-Function Analysis 62) Magnetic Properties
65) Spin-Texture 68) Transport Properties
======================== Misc Utilities =========================
71) Optical Properties 72) Molecular-Dynamics Kit
74) User Interface 78) VASP2other Interface
91) Semiconductor Kit 92) 2D-Material Kit
0) Quit
------------>>
3
===================== K-Path Options ============================
301) 1D Structure
302) 2D Structure
303) Bulk Structure
304) K-Path for Wannier90 Code
305) K-Path for Phonopy Code
306) K-Path for CP2K Code
309) Visualize K-Path in First Brillouin Zone
0) Quit
9) Back
------------>>
303
+---------------------------- Tip ------------------------------+
See An Example in vaspkit/examples/seek_kpath/GaAs_bulk.
The suggested K-Path is only for the standardized primtive cell.
+---------------------------------------------------------------+
-->> (01) Reading Structure from POSCAR File...
+-------------------------- Summary ----------------------------+
Prototype: ABC2
Total Atoms in Input Cell: 8
Lattice Constants in Input Cell: 3.205 3.205 11.264
Lattice Angles in Input Cell: 90.000 90.000 120.000
Total Atoms in Primitive Cell: 8
Lattice Constants in Primitive Cell: 3.205 3.205 11.264
Lattice Angles in Primitive Cell: 90.000 90.000 120.000
Crystal System: Hexagonal
Crystal Class: 6/mmm
Bravais Lattice: hP
Extended Bravais Lattice: hP2
Space Group Number: 194
Point Group: 27 [ D6h ]
International: P6_3/mmc
Symmetry Operations: 24
Suggested K-Path: (shown in the next line)
[ GAMMA-M-K-GAMMA-A-L-H-A|L-M|H-K ]
+---------------------------------------------------------------+
-->> (02) Written HIGH_SYMMETRY_POINTS File for Reference.
-->> (03) Written PRIMCELL.vasp file.
-->> (04) Written KPATH.in File for Band-Structure Calculation.
o----------------------------WARNING----------------------------o
| Do NOT forget to copy PRIMCELL.vasp to POSCAR unless you know |
| what you are doing. Otherwise you might get wrong results! |
o---------------------------------------------------------------o
-->> (05) Written INCAR file!
+-------------------------- Summary ----------------------------+
POTCAR Type: PBE
Number of Elements: 3
POTCAR File No.1: [ POTCAR_Ti_sv ], Valence Electron: 12.0
POTCAR File No.2: [ POTCAR_C ], Valence Electron: 4.0
POTCAR File No.3: [ POTCAR_S ], Valence Electron: 6.0
Total Atoms: 8
Total Valence Electrons: 68.0
+---------------------------------------------------------------+
-->> (06) Written POTCAR File with the Recommended Potential!
注意:将PRIMCELL.vasp 改名为 POSCAR文件,将KPATH.in改名为KPOINTS文件
- 用于能带非自洽计算的INCAR文件模板
ALGO = Normal
EDIFF = 1e-05
ENCUT = 520
IBRION = -1
ISIF = 3
# ISMEAR = -5
ISMEAR = 0
ISPIN = 2
ICHARG = 11
KPAR = 8
LAECHG = True
LASPH = True
# LCHARG = True
LCHARG = .FALSE.
LORBIT = 11
LREAL = Auto
LVHAR = True
LWAVE = False
MAGMOM = 8*-0.0
NELM = 100
NPAR = 4
NSW = 0
PREC = Accurate
SIGMA = 0.05
SYSTEM = Mp-3732
注意:相比静态自洽计算,计算能带的时候,以下几个关键词参数值需要进行修改
ISMEAR = 0
LCHARG = .FALSE.
ICHARG = 11
- vasp能带计算完成后输出文件目录
.
├── AECCAR0
├── AECCAR1
├── AECCAR2
├── CHG
├── CHGCAR
├── CONTCAR
├── DOSCAR
├── EIGENVAL
├── INCAR
├── KPOINTS
├── LOCPOT
├── OSZICAR
├── OUTCAR
├── PCDAT
├── POSCAR
├── POTCAR
├── PROCAR
├── REPORT
├── slurm-2675659.out
├── sub.sh
├── vasprun.xml
├── WAVECAR
└── XDATCAR
- 操作流程
vaspkit --> 21 --> 211
- 输入文件和输出文件
输入文件:INCAR,DOSCAR,POSCAR,EIGENVAL 和 KPOINTS
-->> (01) Reading Input Parameters From INCAR File...
+---------------------------------------------------------------+
| >>> The Fermi Energy will be set to zero eV <<< |
+---------------------------------------------------------------+
-->> (02) Reading Fermi-Energy from DOSCAR File...
-->> (03) Reading Structure from POSCAR File...
-->> (04) Reading Energy-Levels From EIGENVAL File...
-->> (05) Reading K-Path From KPOINTS File...
输出文件:KLABELS,BAND.dat,REFORMATTED_BAND_UP/DW.dat 和 KLINES.dat
-->> (06) Written KLABELS File!
-->> (07) Written BAND.dat File!
-->> (08) Written REFORMATTED_BAND_UP/DW.dat Files!
-->> (09) Written KLINES.dat File!
- KLABELS文件内容
K-Label Coordinate of high-symmetry k-point in band-structure plots
GAMMA 0.000
M 1.132
K 1.786
GAMMA 3.093
A 3.371
L 4.503
H 5.157
A|L 6.464
M|H 6.743
K 7.022
* Tip: Please label each high symmetry point in KPOINTS (KPATH.in) file. Otherwise, they will be identified as 'XX' in KLABELS file
- BAND_GAP
Spin Channel: <UP> <DOWN> <TOTAL>
Band Gap (eV): 0.0009 0.0009 0.0009
Eigenvalue of VBM (eV): 6.1956 6.1956 6.1956
Eigenvalue of CBM (eV): 6.1965 6.1965 6.1965
Fermi Energy (eV): 6.1958 6.1958 6.1958
Highest-Occupied Band: 20 20 20
Lowest-Unoccupied Band: 20 20 20
Location of VBM (UP): 0.0263 0.0000 0.5000
Location of CBM (UP): 0.0175 0.0175 0.5000
Location of VBM (DW): 0.0263 0.0000 0.5000
Location of CBM (DW): 0.0175 0.0175 0.5000
Location of VBM (TO): 0.0263 0.0000 0.5000
Location of CBM (TO): 0.0175 0.0175 0.5000
** Notice: A tiny gap implies your system might be metallic. **
- origin绘图
将BAND.dat文件拖入到origin中,设置横坐标的刻度和标签,如下所示。横坐标的范围和K点信息可以参考KLABELS文件中的内容
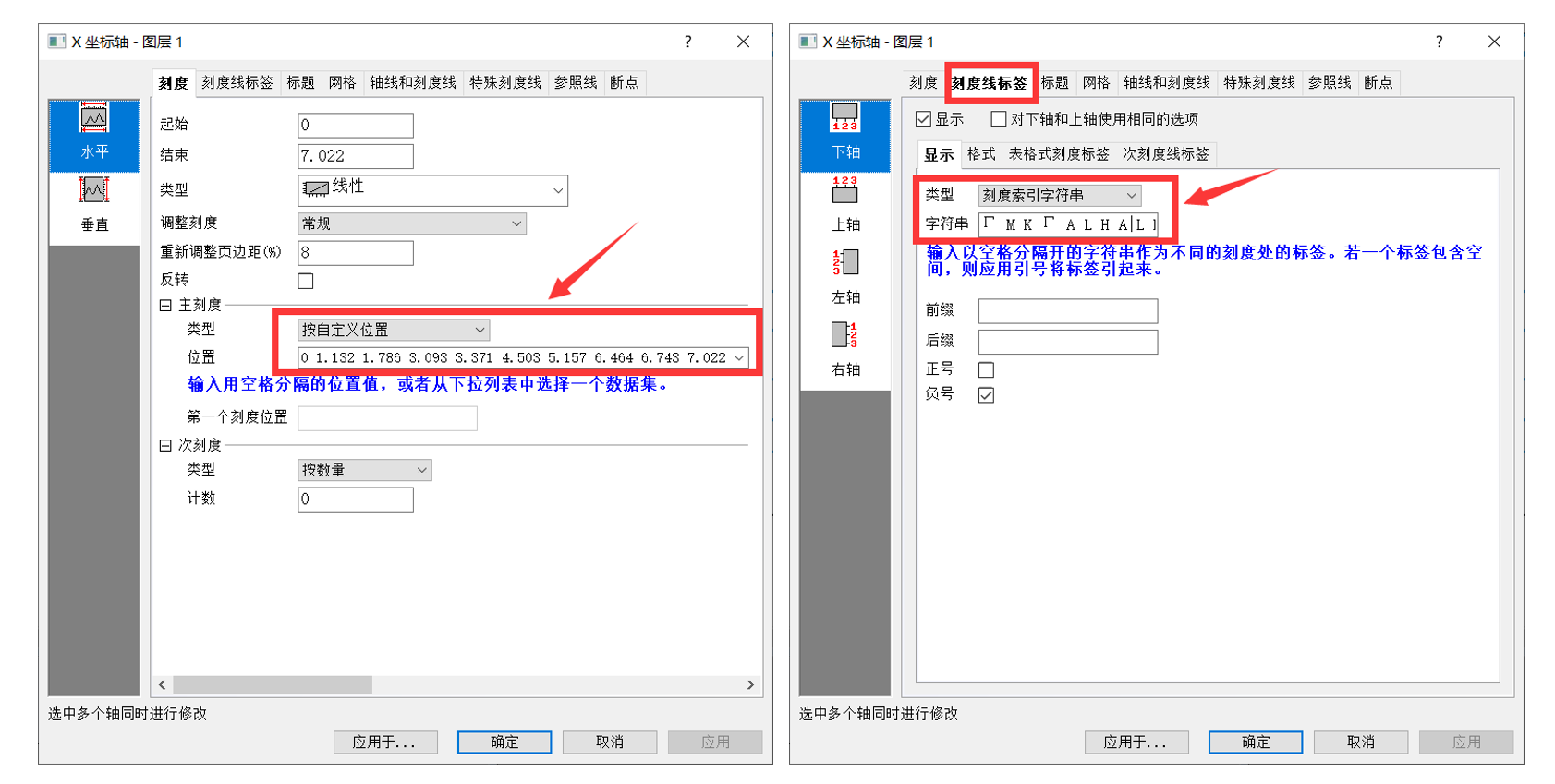
- DFT能带计算:http://vaspkit.cn/index.php/27.html
- VASP+vaspkit计算能带结构+态密度:https://zhuanlan.zhihu.com/p/526969630
- VASP vaspkit Origin 画能带图:https://zhuanlan.zhihu.com/p/564983969
- vaspkit新手必读:http://vaspkit.cn/index.php/66.html
- vaspkit中文官方文档(0.73版本):https://tamaswells.github.io/VASPKIT_manual/manual0.73/vaspkit-manual-0.73.html#header-n402
- vaspkit英文tutorial:https://vaspkit.com/tutorials.html#band-structure
- 大师兄科研网:https://lvaspthw.readthedocs.io/en/latest/LVASPTHW/Ex_06/
- 大师兄科研网:https://www.bigbrosci.com/2018/02/07/ex38/#disqus_thread