5. Denoising quick guide! - VascoElbrecht/JAMP GitHub Wiki
JAMP comes with a denoising module Denoise() which can be used to extract haplotype sequences from metabarcoding datasets (See Elbrecht et al. 2018). Instead of clustering reads and obtaining centroids, sequences are denoised using Unoise3 followed by additional abundance based filtering steps (Figure 1).
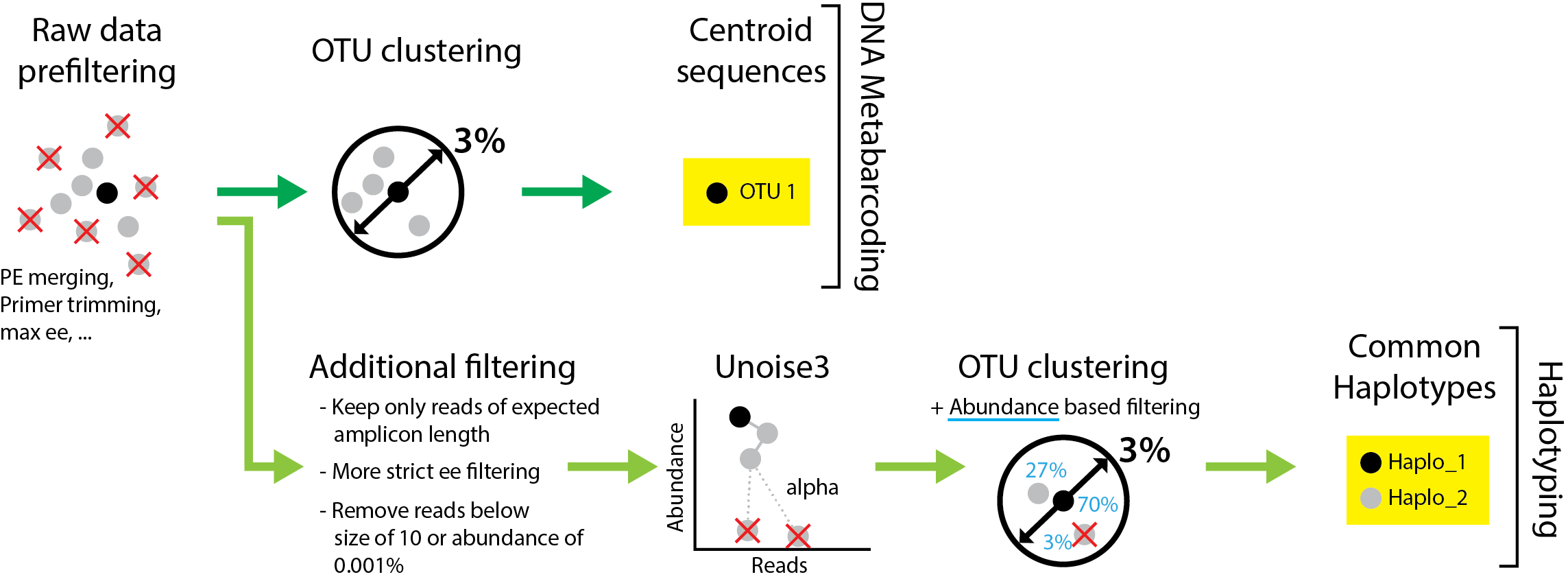 Figure 1 Comparison of commonly used clustering based sequence processing (generating only one centroid sequence per OTU) with the denoising based approach implemented in JAMP, which can extract the common haplotypes from the dataset.
Figure 1 Comparison of commonly used clustering based sequence processing (generating only one centroid sequence per OTU) with the denoising based approach implemented in JAMP, which can extract the common haplotypes from the dataset.
A) Read Prefiltering
The haplotyping approach implemented in JAMP can use the same prefiltered data sets which are used for OTU clustering in common metabarcoding pipelines. However, two additional filtering steps are recommended: 1) More stringent error filtering can be useful to further reduce false positive sequences (U_max_ee(max_ee=0.2)). 2) All reads longer or shorter than the expected amplicon length should be discarded, as they likely represent sequences affected by insertions or deletions (Minmax(min=421, max=421)). This means that this approach can only be applied on coding genes (not ribosomal), and even there only taxa with the common amplicon length can be detected (e.g. will also discard true sequences of taxa which have an codon insertion or deletion).
The input files for the Denoise() command are a single undereplicated fasta files for each sample. The Denoise() function will apply all additional denoising and filtering steps. To remove decrease noise in the data, within each sample reads are dereplicated and low abundant reads are discarded. The minimum number of each sample to keep can be modified with minsize = 10, and the relative abundance minrelsize = 0.001 % is used as well. This can be useful if sequencing depth does vary between samples, however, it's generally recommended that all samples have the same sequencing depth (as denoising is applied on pooled sequences and no sample should be overrepresented). Thus, before Denoising samples can be adjusted to the same read numbers using U_subset().
B) Read denoising
Next, Prefiltered reads are pooled into one sample, dereplicated and denoised using the unoise3 algorithm (Edgar 2016). This is based on the difference in abundance between similar sequences, under the assumption that sequences that are generated by sequencing errors are less abundant than true sequences. This of cause only is true for abundant sequences, as low abundant true sequences will get lost in the see of noisy sequences (thus we apply additional threshold-based filtering at a later stage!). The distance between sequences, at which the low abundant one is discarded is defined by the alpha value (see figure 1). The lower the alpha value the more strict the filtering! a Unoise_alpha=5 should be good in most cases, but this might vary based on the data sets and organisms.
Future Denoise() version might feature a selection of different denoising algorithms, but at this stage, only unoise3 is available (strategy = "unoise").
C) OTU clustering + haplotype table
After read denoising, many sequences affected by errors should have been removed. However, not all true haplotypes can be removed from the sequencing noise, especially the low abundant ones. Here additional filtering is required. Also, haplotypes which did remain after read-denoising should be grouped together by taxon, so the results can be analyzed further.
Thus read clustering using Usearch is applied (cluster_otus) to the denoised reads using 3% sequence similarity threshold, followed by exact matching all samples against the resulting OTU list. The resulting haplotype table gives information on which haplotypes are present in which samples, and how they group together into OTUs (Raw_haplotable.csv).
D) Additional threshold based filtering
The raw haplotpye table is the basis for the next threshold-based filtering steps. All OTUs below OTUmin = 0.01% relative abundance in at least one sample are discarded. Further, all haplotypes below minhaplosize=0.003% relative abundance in at least one sample are discarded. This removes low abundant OTUs likely affected by noise, and also rare haplotypes across the dataset which might be sequences affected by artifacts, despite sequence denoising.
To increase the reliability of the results haplotypes within each OTU should be additionally filtered (rather ten discarding low abundant haplotypes more aggressively using minhaplosize. Thus within each OTU for each individual sample haplotypes below e.g. 5% relative abundance can be set to 0 abundance with withinOTU = 5. It has to be stressed that this filtering step will remove a large proportion of correct haplotypes, but also decreases the probability to include sequences affected by errors in the analysis. This filtering step might not be appropriate for all analysis, but helpful if e.g. haplotype networks or maps should be generated (to observe the main patterns abundant haplotypes are sufficient).
Cut off filtering will always lead to edge effects. However, OTUs which have very little abundance in certain samples could be more severely affected by edge effects and are generally more likely derived from noise in the data. While low abundant OTUs are already discarded globally with OTUmin = 0.01 and rare haplotypes are removed with minrelsize=0.001, additionally (within individual samples) discarding OTUs with a sequence abundance of e.g. below 200 can further reduce noise in the data. This can be done with eachsampleOTUmin = 200 but is usually deactivated eachsampleOTUmin = NULL
Additionally, for data sets with many samples (e.g. more than 30), presence-based filtering can be useful. This can be done by discarding haplotypes which are not present in at least minHaploPresence=1 sample, or similarly OTUs which are not present in at least minOTUPresence=1 samples. Both values are set to 1 by default (meaning that all OTUs and haplotypes will remain in the dataset).
Tipp
With the grep pattern defined in renameSamples="(.*_.*)_cut.*") the sample names in the output tables can be changed. This can be used so sample names are containing only the sample ID instead of the processing history added in the file names when using the JAMP pipeline.
Output files
The Denoise() function produces many output files! Usually, you want to use the haplotype tables E_haplo_table.csvor E_haplo_table_rel.csv (relative abundance) in the base directory and the fasta file _data/4_denoised/E_haplo_sequ.txt or _data/4_denoised/E_haplo_sequ.txt for fasta files containing the haplotype sequences sorted by OTU or haplotype number.
Plase, notice that the haplotypes are clustered by 3% similarity into OTUs so one can figure out which belong together.
Here is an explanation of additional files created in _data:
1_derep- As unoise3 discards abundance information, and to make sure all samples use the same haplotype names, sequences are renamed in this step to retain this information (renamed files saved in2_renamed).3_unoise- Sequences that are discarded as part of the unoise3 step are also removed from the individual files, which are then matched to the haplotype list to generate a haplotype table (similar to an OTU table). This haplotype list is also clustered with 3% similarity to identify which haplotypes belong together.4_denoised- Next, the haplotype table is generated and subjected to several abundance based filtering steps.
Additionally, statistics and plots are provided in the _stats folder:
1_derep_logs.txtand2_unoise.txt- unoise / vsearch logsE_Haplotype_presence.pdf- Histogram showing the presence of haplotpyes across samples (and cut offminHaploPresence=1)E_OTU_presence_across_samples.pdf- Plot showing the presence of OTUs across samples (and cut offminOTUPresence=1)