Practical Section 1: Quality control and pre processing - TarifenoLab/Hands-on-RNA-seq-Data-Analysis GitHub Wiki
To start to understand the data we will explore the quality of sequencing. This procedure must be done before any further analysis and it is known as Reads Pre-Procesing
In this analysis, we will use the programs FastQC and Trimmomatic the following links will drive you to the program pages. The secret to success in bioinformatics is that review the manual and fully understand what is the program doing, this will help you to be independent in your analysis and also (very important) understand the results.
Link to FastQC manual (ShortCut: Check sections Documentation and Example Reports)
Link to Trimmomatics manual XD (ShortCut: Check first 4 pages)
-
Go to the Tools Menu (Left side of your screen) and select
- GENOMIC FILE MANIPULATION --> FASTQ Quality Control --> FastQC Read Quality reports
-
Select the button “multiple datasets”, then click on "browse" and select all the data sets then press "OK". By doing this, we will run the analysis on batch mode, which means that all files will be analyzed separately and give separate outputs.
-
Scroll down leaving all other options as default and click on the button "Execute"
-
Calculating the QC report can take some minutes, the results will appear on your history.
-
For each file, we will have two reports:
-
RawData: This is are the raw quality metrics. -
WebPage: This is the report to visualize the metrics calculated on the RawData.
-
Step 2. We will review the report for one replicate of the WT condition and the MUT condition
- Just remember that you have two files by sample (R1 and R2). To do so, just click on the "Eye icon"
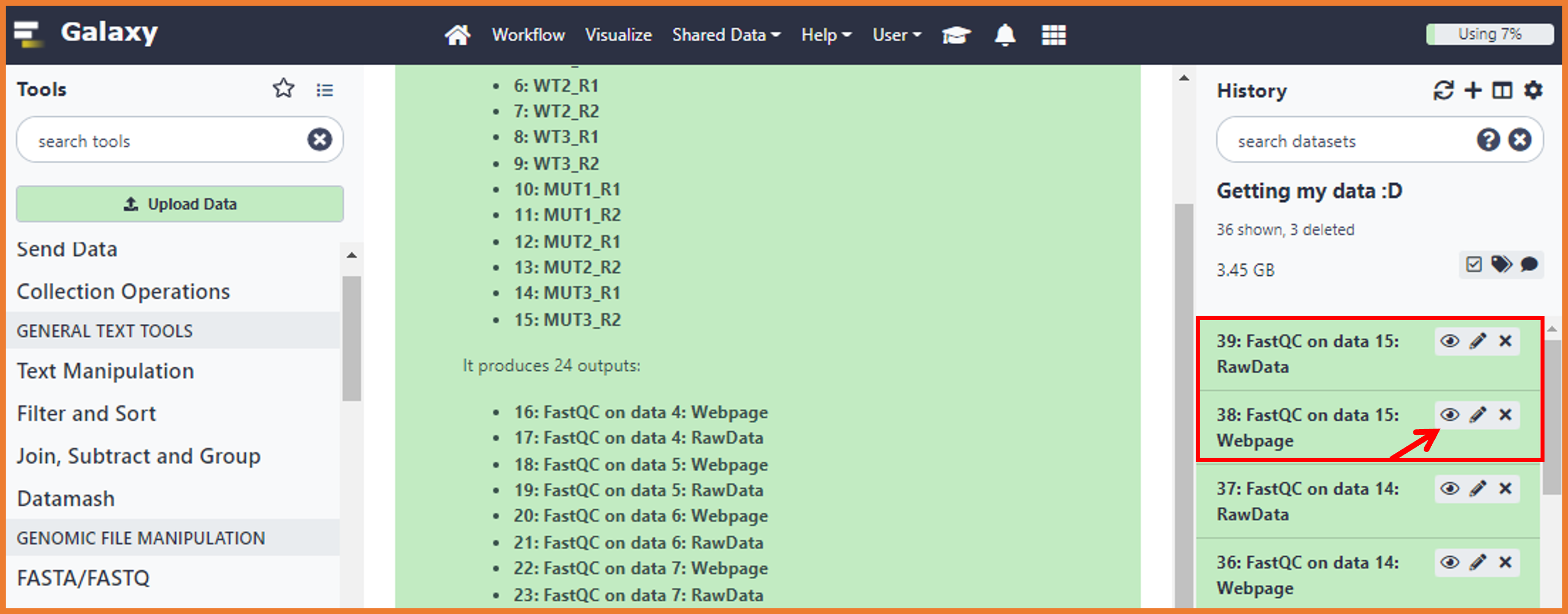
-
The report will score the quality either as a PASS (OK), WARNING (Maybe wrong, please check), or Fail (wrong). Depending on the aspect evaluated, based on the report, we will decide about the pre-processing approach to remove bad quality data improving the performance of the further analysis. It is important to notice that this QC report is done for different NGS approaches, so, some of the aspects evaluated here might not be applicable or important for RNA-seq data. In this sense, we will focus on the following aspects:
- Basic Statistics
- Per base sequence quality
- Per sequence quality scores
- Per base sequence content
- Per base N content
- Sequence Length Distribution
- Sequence Duplication Levels
- Overrepresented sequences
- Adapter Content
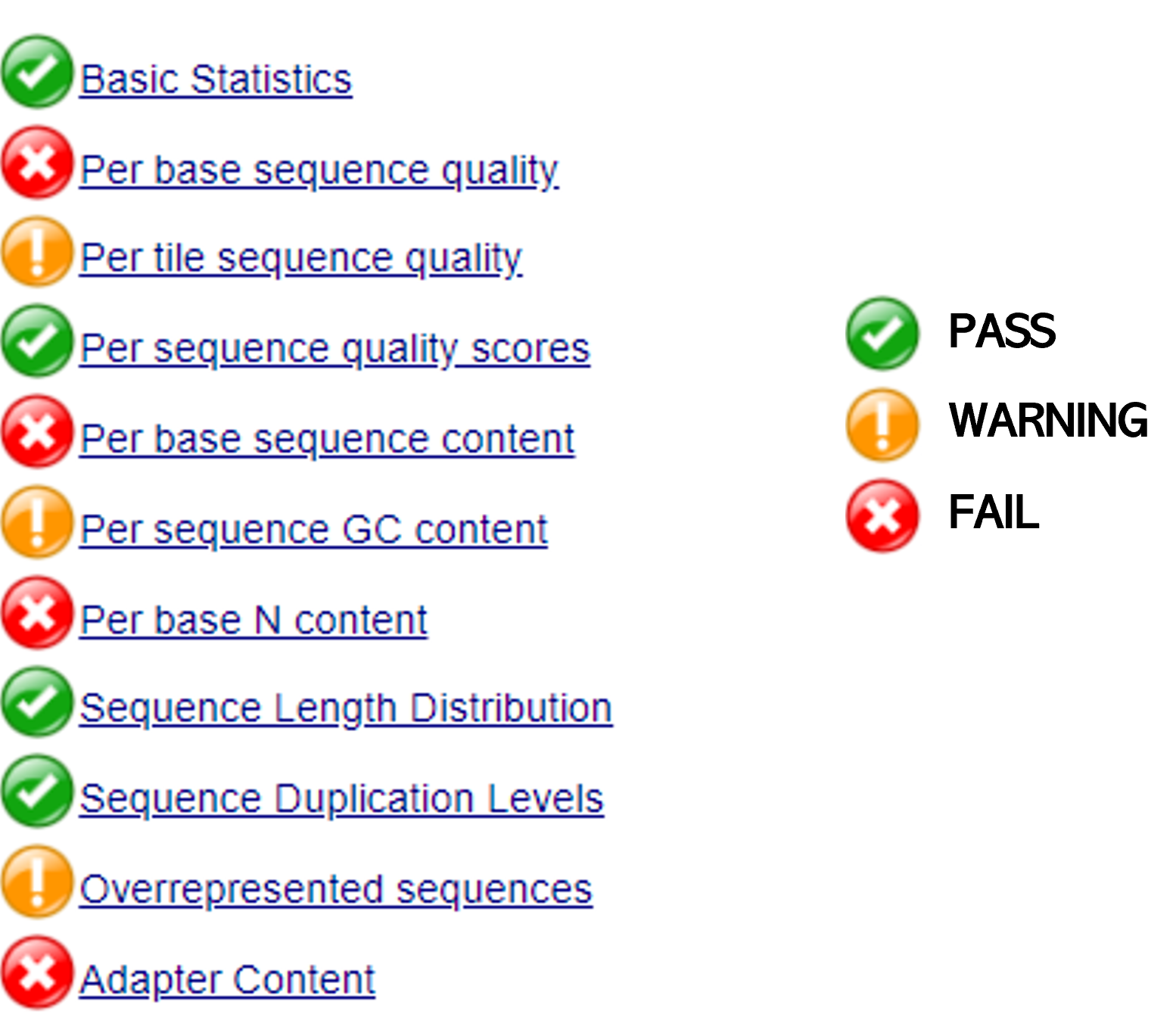
- To interpret your results, go back to the
FastQCmanual (shortCut: check the "Analysis Module" section)
Instructions
- Makes Groups of 3 persons
- Discuss with your group the 3 aspects that will be designed by the Professor
- You will have 15-20 minutes to elaborate a 3 slides presentation (one by aspect) explaining the results and proposing the pre-processing approach
- Add your slides to the collaborative ppt hoster here, this collaborative presentation will be used for the final discussion.
In this step, and just for simplicity, we will focus on removing bad quality nucleotides and search for adaptors that we know are present on the libraries (Nextera library preparation protocol).
🚨🚨🚨
However, it is important to notice that several quality parameters can affect the quality of your data and they must be assessed before the next analysis step (the reads mapping). For example, any custom adapter, sequences that after pre-processing are too short, contaminant sequences, etc... Just keep in mind that in bioinformatics the rule "garbage in -- garbage out" really applies!!
🚨🚨🚨
-
Go to the Tools Menu (Left side of your screen) and select
- GENOMIC FILE MANIPULATION --> FASTQ Quality Control --> Trimmomatics
-
Set the following options
-
Single-end or paired-end reads?
- Paired-end (two separate input files)
-
Input FASTQ file (R1/first of pair)
- Select the R1 of a sample (e.i. WT1_R1)
-
Input FASTQ file (R2/second of pair)
- Select the R2 of a sample (e.i. WT1_R2)
As you can see, the pre-processing analysis is performed by samples including both reads. This means that we will have to run the 6 jobs (one by samples).
-
-
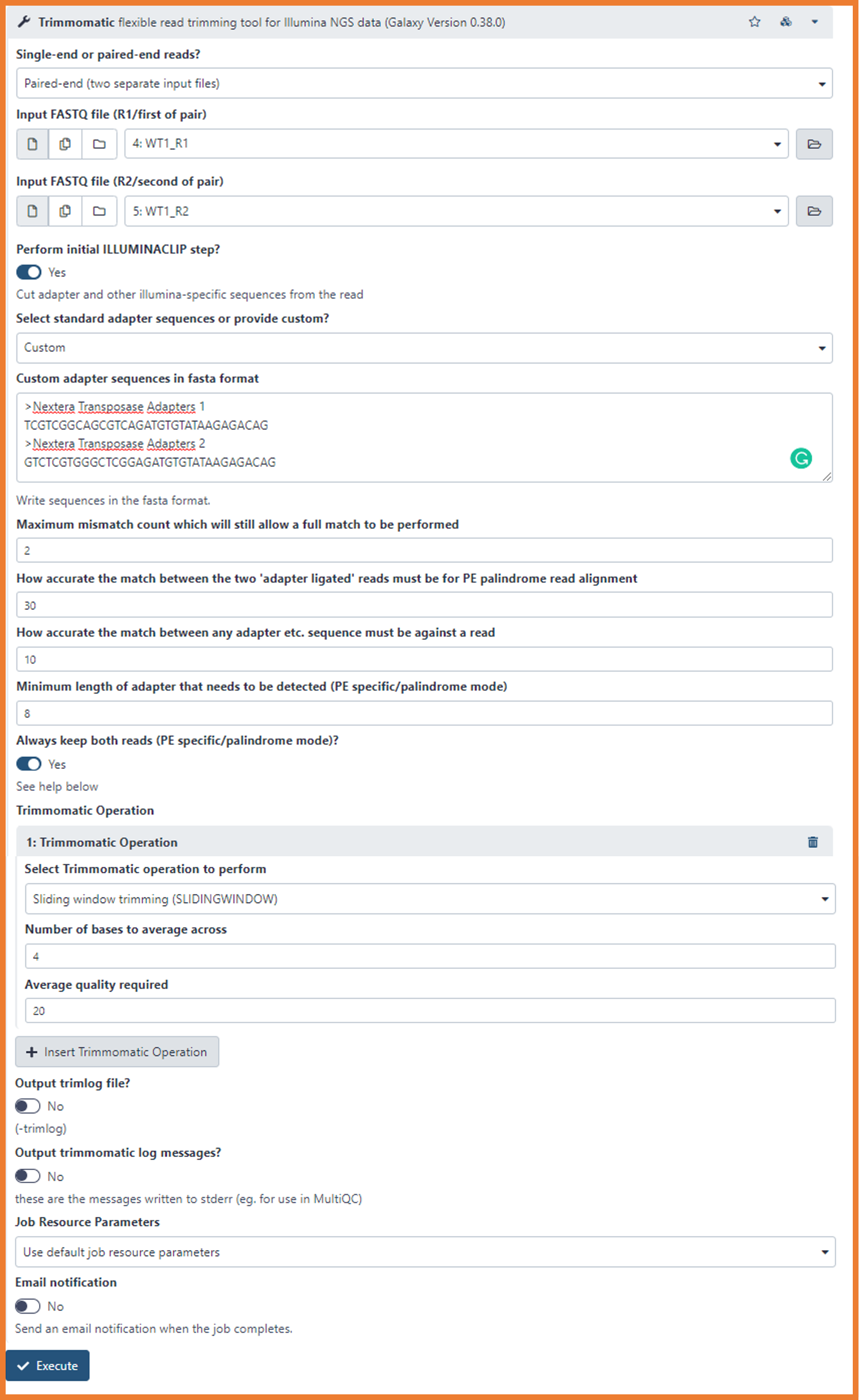
First, let's set the adapter clipping parameters. On our case, we will perform a custom clipping providing the sequences that we want to remove.
- Perform initial ILLUMINACLIP step?
- Turn on the button to yes
- Select standard adapter sequences or provide custom?
- Nextera (paired-ended)
- Leave the following parameters associated with adapter clipping in default:
Maximum mismatch count which will still allow a full match to be performed How accurate the match between the two 'adapter ligated' reads must be for PE palindrome read alignment
How accurate the match between any adapter etc. sequence must be against a read
Minimum length of adapter that needs to be detected (PE specific/palindrome mode)
Always keep both reads (PE specific/palindrome mode)?
- Perform initial ILLUMINACLIP step?
-
Now, we will select the trimming options to remove bad quality nucleotides
- Select Trimmomatic operation to perform
- Sliding Window trimming (SLIDINGWINDOW)
- Number of bases to average across
- 4
- Average quality required
- 25
- Run the pre-processing by clicking on "Execute"
- Select Trimmomatic operation to perform
Trimommatics will produce 4 outputs files as follow (example using sample MUT3_R1 and MUT3_R2):
- Trimmomatic on MUT3_R1 (R1 paired)
- Trimmomatic on MUT3_R2 (R2 paired)
- Trimmomatic on MUT3_R1 (R1 unpaired)
- Trimmomatic on MUT3_R2 (R2 unpaired)
The "unpaired" files contains reads that due to the preprocessing have lost their mates. This sequences will not be included on the further analysis, so, we will remove them from our history clicking the icon "X". Depending on the load of the Galaxy server, it can takes some minutes to get all the results. In the meantime, we will perform the next step with the results you already got!
FastQC MUT1_R1 https://usegalaxy.org/datasets/bbd44e69cb8906b520a348f58af237dd/display?to_ext=fastqsanger
FastQC MUT1_R2 https://usegalaxy.org/datasets/bbd44e69cb8906b5b079ae7dae41c013/display?to_ext=fastqsanger
Once the pre-processing is done, we need to evaluate if the quality of the sequences has improved. To do so, we need to run again the program FastQCon the pre-processed samples.
- Here, I leave you an example of the before and after of the sample MUT1_R1 concerning the "Per base sequence quality"
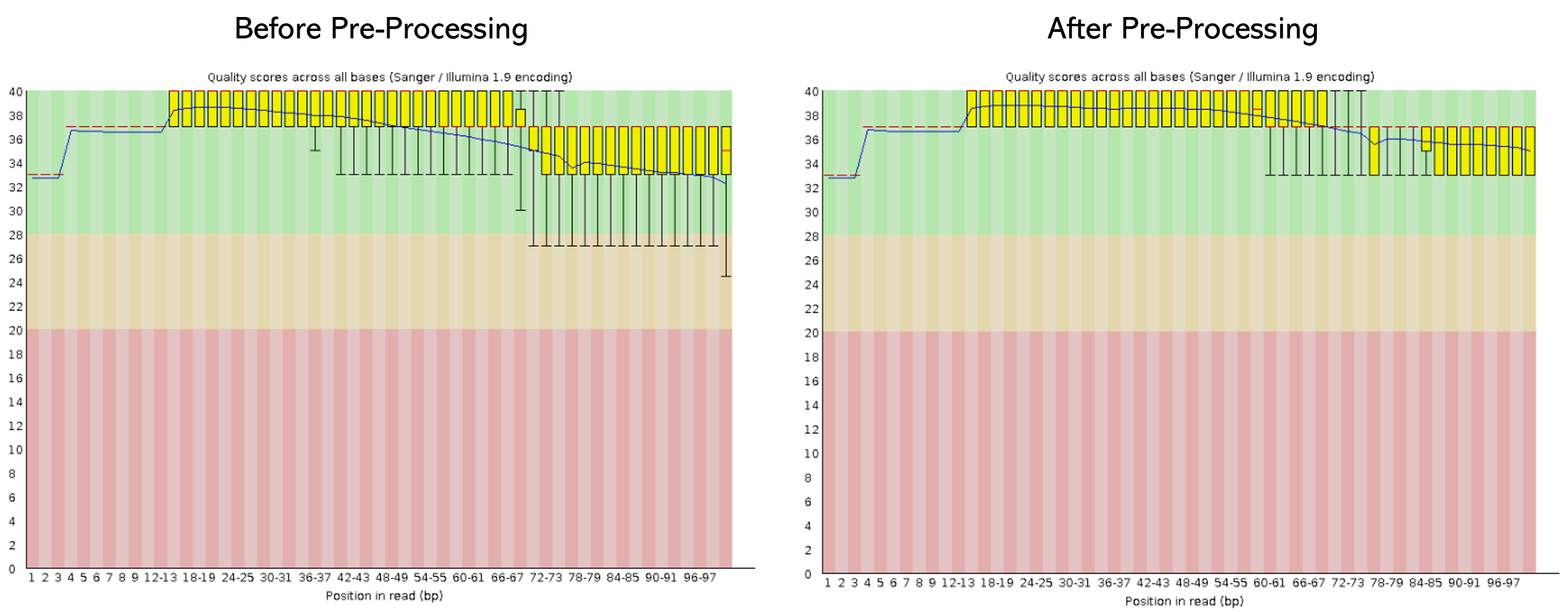
- So, let´s check the quality of one MUT and WT sample (for R1 and R2)
Instructions
- In Groups of 3 persons discussed the results obtained by the QC analysis
With the last results, we finish Section 1: Quality control and pre-processing 💻💻