Task 4: ATAC seq analysis for the ENCDO451RUA ENTEx project donor - PatiGenius/EpigenomicsTask_MscOmics GitHub Wiki
ATAC-seq analysis
We perform an ATAC-seq analysis with the aim to obtain the number of peaks that fall outside the gene coordinates (chromatin-accessibility signatures). These peaks will be used in task 5 to perform the distal regulatory elements catalogue.
We move to folder ATAC-seq, and create folders to store bigBed data files and peaks analyses files. We organise them in a consistent way (as done for ChIP-seq)
We start from epigenomics_uvic directory. We move to the created ATAC-seq folder and generate the new folders.
cd ATAC-seq
mkdir analyses
mkdir analyses/peaks.analyses
mkdir annotation
mkdir data
mkdir data/bigBed.files
mkdir data/bed.files
We retrieve from a newly generated metadata file ATAC-seq peaks (bigBed narrow, pseudoreplicated peaks, assembly GRCh38) for stomach and sigmoid colon for the donor ENCDO451RUA.
- First, we need to obtain the link that allows us to obtain the metadata file. From ENCODE project web, we look for the ATAC-seq information belonging to the ENCD0451RUA donor, both for stomach and sigmoid colon tissues and GRCh38 assembly.
We obtain a .txt file that contains the following link: “https://www.encodeproject.org/metadata/?type=Experiment&replicates.library.biosample.donor.uuid=d370683e-81e7-473f-8475-7716d027849b&status=released&assay_title=ATAC-seq&biosample_ontology.term_name=sigmoid+colon&biosample_ontology.term_name=stomach”
We download the metadata.tsv using the next code:
../bin/download.metadata.sh “https://www.encodeproject.org/metadata/?type=Experiment&replicates.library.biosample.donor.uuid=d370683e-81e7-473f-8475-7716d027849b&status=released&assay_title=ATAC-seq&biosample_ontology.term_name=sigmoid+colon&biosample_ontology.term_name=stomach”
- Second, we are now able to filter for the information we want:
grep -F "bigBed_narrow" metadata.tsv | grep -F "pseudoreplicated_peaks" | grep -F "GRCh38" | awk "BEGIN{FS=OFS="\t"}{print $1, $10, $22}" | sort -k2,2 -k1,1r | sort -k2,2 -u > analyses/bigBed.peaks.ids.txt
We obtain the following ids:
-
ENCFF28TUHP for sigmoid colon
-
ENCFF762IFP for stomach
- At this point, we have the ids and can obtain the bigBed files:
cut -f1 analyses/bigBed.peaks.ids.txt | while read filename; do wget -P data/bigBed.files "https://www.encodeproject.org/files/$filename/@@download/$filename.bigBed" done

- We test whether the msd5 values match with the ones obtained:
../bin/selectRows.sh <(cut -f1 analyses/"$file_type".*.ids.txt) metadata.tsv | cut -f1,45 > data/"$file_type".files/md5sum.txt cat data/"$file_type".files/md5sum.txt | while read filename original_md5sum; do md5sum data/"$file_type".files/"$filename"."$file_type" | awk -v filename="$filename" -v original_md5sum="$original_md5sum" 'BEGIN{FS=" "; OFS="\t"}{print filename, original_md5sum, $1}' done > tmp mv tmp data/"$file_type".files/md5sum.txt awk '$2!=$3' data/"$file_type".files/md5sum.txt done
We obtain the md5sum.txt file, stored in data folder, and we can see the following names:
less data/bigBed.files/md5sum.txt
The names correspond to the ones stored in the bigBed.peaks.ids.txt
- We transform the data to bedFiles:
cut -f1 analyses/bigBed.peaks.ids.txt | while read filename; do bigBedToBed data/bigBed.files/"$filename".bigBed data/bed.files/"$filename".bed done
At this point, we can work with the bed files.
For each tissue, we run an intersection analysis using BEDTools and report the following:
To solve this section, we have to take in mind the next figure:
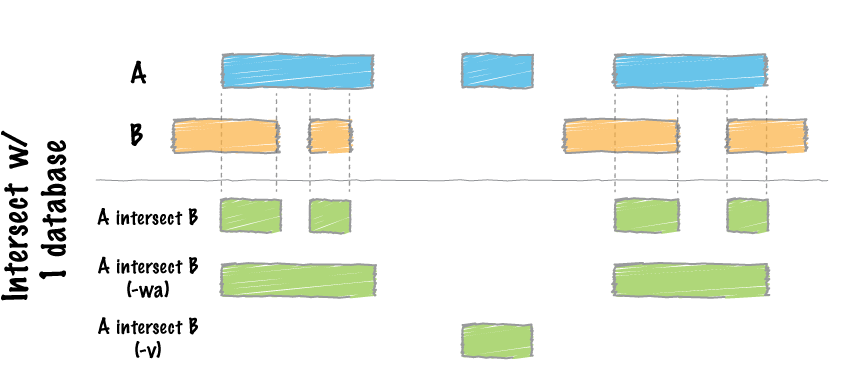
Where we intersect two data bases, A and B, and retrieve the peaks from A that overlap B. We can use different arguments in the function as for example "-u", "-wa", "-wb" and -"u", which allow us to obtain the information in different ways.
The number of peaks that intersect promoter regions
The data query (A: -a) corresponds to the bed file format available for each one of the tissues. From this data, we want to retrieve the number of peaks that intersect promoter regions. Coordinates of promoter regions are stored in the gencode.v24.protein.coding.non.redundant.TSS.bed database (B, -b).
cat analyses/bigBed.peaks.ids.txt | while read filename tissue; do bedtools intersect -a data/bed.files/"$filename".bed -b annotation/gencode.v24.protein.coding.non.redundant.TSS.bed -u | cut -f7 | sort -u > analyses/peaks.analyses/genes.with.peaks."$tissue".txt done
The total number of peaks that intersect promoter regions in each tissue is:
- Sigmoid colon: 38071 peaks that intersect promoter regions

- Stomach: 33169 peaks that intersect promoter regions

The number of peaks that fall outside gene coordinates (whole gene body).
The data query (A: -a) corresponds to the one in bed file format for each one of the tissues. From this data, we want to retrieve the number of peaks that fall outside gene coordinates. Whole gene body coordinates are stored in the database gencode.v24.protein.coding.gene.body.bed (B, -b). We need to intersect both data bases and retrieve the regions that do not overlap ("-u").
We follow 2 steps:
- We need to obtain the gene coordinates for the whole gene body from the primary assembly file (gencode.v24.primary_assembly.annotation.gtf)
We did it in class, in the ChIP-seq analysis, so we just need to move the file from the annotation folder that belongs to ChIP-seq analysis to the annotation folder in ATAC-seq analysis directory.
cp ../ChIP-seq/annotation/gencode.v24.protein.coding.gene.body.bed annotation
- Now, we can look for the number of peaks that fall outside gene coordinates by computing the next:
We remove the "-u" because it writes the original A entry once if any overlaps found in B, and what we want to obtain is the opposite, the original A entry that do not overlap B. We indicate it with the "-v".
cat analyses/bigBed.peaks.ids.txt | while read filename tissue; do bedtools intersect -a data/bed.files/"$filename".bed -b annotation/gencode.v24.protein.coding.gene.body.bed -v | sort > analyses/peaks.analyses/genes."$tissue".outside.gene.coord.txt done
- For stomach: 34537 peaks that fall outside gene coordinates.

- For sigmoid colon: 37035 peaks that fall outside gene coordinates.

We save both files in peaks.analyses folder within analyses directory.
At this point, we have obtained the peaks that fall outside gene coordinates, for the whole gene body and for both stomach and sigmoid colon tissues. We can use this data for the following exercise, Task 5.