Disease Module Overlap - PathwayAnalysisPlatform/ProteoformNetworks GitHub Wiki
We create a gene interaction network from a pathway database (Reactome[1]). Genes are the nodes and the links connect genes whose products participate in the same reactions accross the pathways.
Having gene sets related to diseases, we can delimit subnetworks by selecting nodes corresponding to genes in the set along with its connecting links. Each of this subnetworks are disease modules.
There are cases when two disease modules overlap (share common nodes) suggesting similarities between the diseases at molecular level. In this analysis we verify what happens when we convert gene nodes into proteoform nodes as a proteoform interaction network also created from the reference pathway database.
- Comparison of modules accross levels:
- Module sizes, variation and percentages
- Connection density
- Module topology metrics
- Comparison of overlap scores
- Overlap coefficient: Values, distribution
- Size variation vs score
- Selected examples with certain overlap size
- Read entities: genes, proteins and proteoforms
- Get disease gene sets
- Create disease modules
- Convert modules to proteoform modules
- Discard disconnected proteoforms from modules
- Find pairs of overlapping diseases
- Calculate overlapping score for each overlapping pair at gene and proteoform level
- Make a distribution plot of all overlapping scores at gene and proteoform level
- Get the disease module pairs that got the biggest reduction in overlapping score
- Get the disease module pairs that got the biggest increase in overlapping score
- Check for pairs of overlapping diseases which have modified proteins as overlap
We get the disease to gene associations from PheGenI. This resources contains association results from multiple genome-wide association study (GWAS) where many single nucleotide polimorphisms (SNPs) were found associated to phenotypes (traits). Among those phenotypes, there are diseases.
The full data set can be downloaded from here.
The data is a tab separated file with columns for Trait, P-Value, and Gene Id among others. To create the gene sets, we select only genes which have a SNP associated to a trait withing a cuttoff p-value of 5 x 10-8 for a genome wide significance, in a similar fashion as a reference study[3].
The number of phenotypes considered are 846 with 3292 genes associated to them.
Details on the implementation here.
Dataset statistics:
- Number of traits: 790
- Disease pairs with at least one module containing modified proteoforms: 947418
- Total number of disease pairs: 1378276
- Disease pairs with at least one module containing 90% modified proteins: 4696
The score[3] compares the distances between nodes in the same disease module to the distances between nodes of different modules. Given a pair of disease modules A and B it calculates the average distance between nodes of A and then the distances between nodes in B. Afterwards, it calculates the average distance between each node in A to each node in B. Finally, it calculates the difference between those averages.
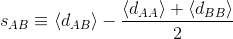
- Get list of genes from the Reactome[1] graph database in NEO4J console:
MATCH (ewas:EntityWithAccessionedSequence{speciesName:'Homo sapiens'})-[:referenceEntity]->(re:ReferenceEntity{databaseName:'UniProt'})
WITH re.identifier as protein, re.geneName as genes
WHERE size(genes) > 0
UNWIND genes as gene
RETURN DISTINCT gene
Note: Delete the header line of the file.
-
Download disease - gene association data from PheGenI[2] from here.
-
Filter records using a genome wide cutoff (p-value < 5 x 10-8). Use the script: src\Python\filter_genes.py
-
Read gene sets for each Trait (disease or Phenotype) from the PheGenI data and filter to only those genes also in the Reactome database.
- Compile:
g++ src/Cpp/main.cpp src/Cpp/overlap.cpp src/Cpp/bimap.cpp src/Cpp/phegeni.cpp src/Cpp/utility.cpp -o Debug/analysis -std=c++17- Execute:
./Debug/analysis.exe
1.1 Download and install Neo4j Community Edition:
https://neo4j.com/download-center/#community
1.2 Extract to the desired location, for example:
C:\Program Files\Neo4j\
1.3 Download Reactome graph database:
https://reactome.org/download/current/reactome.graphdb.tgz
1.4 Extract the contents to the the Neo4j directory:
C:\Program Files\Neo4j\neo4j-community-3.5.12\data\databases
1.5 Edit neo4j.conf file.
- Disable the authentication.
- Enable upgrade from an older version.
- (Optional) Set the correct name of the database, if the name of the graph.db folder was changed.
1.6 Run Neo4j with the command:
C:\Program Files\Neo4j\neo4j-community-3.5.12\bin\neo4j console
1.7 Create gene, protein and proteoform csv files
At the Neo4j browser: http://localhost:7474/browser/
Execute the Cypher queries to get the lists of genes, proteins and protoeforms. Save the result files with the user interface button to "Export CSV" in the project directories:
-
resources/Reactome/Genes/
-
resources/Reactome/Proteins/
-
resources/Reactome/Proteoforms/
-
Genes:
MATCH (ewas:EntityWithAccessionedSequence{speciesName:'Homo sapiens'})-[:referenceEntity]->(re:ReferenceEntity{databaseName:'UniProt'})
WITH re.identifier as protein, re.geneName as genes
WHERE size(genes) > 0
UNWIND genes as gene
RETURN DISTINCT gene
- Proteins:
MATCH (pe:PhysicalEntity{speciesName:"Homo sapiens"})-[:referenceEntity]->(re:ReferenceEntity{databaseName:"UniProt"})
RETURN DISTINCT re.identifier as protein
- Proteoform:
MATCH (pe:PhysicalEntity{speciesName:'Homo sapiens'})-[:referenceEntity]->(re:ReferenceEntity{databaseName:'UniProt'})
WITH DISTINCT pe, re
OPTIONAL MATCH (pe)-[:hasModifiedResidue]->(tm:TranslationalModification)-[:psiMod]->(mod:PsiMod)
WITH DISTINCT pe.stId AS physicalEntity,
re.identifier AS protein,
re.variantIdentifier AS isoform,
tm.coordinate as coordinate,
mod.identifier as type ORDER BY type, coordinate
WITH DISTINCT physicalEntity,
protein,
CASE WHEN isoform IS NOT NULL THEN isoform ELSE protein END as isoform,
COLLECT(type + ":" + CASE WHEN coordinate IS NOT NULL THEN coordinate ELSE "null" END) AS ptms
RETURN DISTINCT isoform, ptms
ORDER BY isoform, ptms
Then convert the proteoform format from NEO4J to SIMPLE. Use PathwayMatcher class called ProteoformFormatConverter.
java -cp PathwayMatcher.jar matcher.tools.ProteoformFormatConverter Reactome/Proteoforms/ all_proteoforms_v72_neo4j.csv all_proteoforms_v72_simple.csv
-
Execute Jupyter notebook called: analysis_disease_module.ipynb
-
Execute main C++ program to create modules and calculate overlaps.
Set the 10 required parameters:
../../../resources/PheGenI/PheGenI_Association_genome_wide_significant.txt
../../../resources/Reactome/genes.tsv
../../../resources/Reactome/proteins.tsv
../../../resources/Reactome/proteoforms.tsv
../../../resources/Reactome/genes_interactions.tsv
../../../resources/Reactome/proteins_interactions.tsv
../../../resources/Reactome/proteoforms_interactions.tsv
../../../resources/UniProt/mapping_proteins_to_genes.tsv
../../../resources/UniProt/mapping_proteins_to_proteoforms.tsv
../../../reports/modules/
- Get members of both pathways at the different levels of granularity. The pathway names, isoforms and post translational modifications responsible for decomposing the gene level only overlap will show up.
Genes:
MATCH (pathway:Pathway{speciesName:"Homo sapiens"})-[:hasEvent*]->(rle:Reaction{speciesName:"Homo sapiens"}),
(rle)-[:input|output|catalystActivity|physicalEntity|regulatedBy|regulator|hasComponent|hasMember|hasCandidate*]->(pe:PhysicalEntity{speciesName:"Homo sapiens"})-[:referenceEntity]->(re:ReferenceEntity{databaseName:'UniProt'})
WITH pathway, rle, re.identifier as protein, re.geneName as genes
WHERE size(genes) > 0 AND pathway.stId IN ["R-HSA-110056", "R-HSA-6783783"]
UNWIND genes as gene
WITH DISTINCT pathway, gene, protein
RETURN DISTINCT collect(DISTINCT pathway.stId), gene
Proteins:
MATCH (pathway:Pathway{speciesName:"Homo sapiens"})-[:hasEvent*]->(rle:Reaction{speciesName:"Homo sapiens"}),
(rle)-[:input|output|catalystActivity|physicalEntity|regulatedBy|regulator|hasComponent|hasMember|hasCandidate*]->(pe:PhysicalEntity{speciesName:"Homo sapiens"})-[:referenceEntity]->(re:ReferenceEntity{databaseName:'UniProt'})
WHERE pathway.stId IN ["R-HSA-110056", "R-HSA-6783783"]
RETURN DISTINCT collect(DISTINCT pathway.stId), re.identifier
Proteoforms:
MATCH (pathway:Pathway{speciesName:"Homo sapiens"})-[:hasEvent*]->(rle:Reaction{speciesName:"Homo sapiens"}),
(rle)-[:input|output|catalystActivity|physicalEntity|regulatedBy|regulator|hasComponent|hasMember|hasCandidate*]->(pe:PhysicalEntity{speciesName:"Homo sapiens"})-[:referenceEntity]->(re:ReferenceEntity{databaseName:'UniProt'})
WHERE pathway.stId IN ["R-HSA-109703", "R-HSA-111447"]
WITH DISTINCT pathway, rle, pe, re
OPTIONAL MATCH (pe)-[:hasModifiedResidue]->(tm:TranslationalModification)-[:psiMod]->(mod:PsiMod)
WITH DISTINCT pathway, rle.stId as reaction, pe.stId AS physicalEntity,
re.identifier AS protein, re.variantIdentifier AS isoform, tm.coordinate as coordinate,
mod.identifier as type
ORDER BY type, coordinate
WITH DISTINCT pathway, reaction, physicalEntity, protein,
CASE WHEN isoform IS NOT NULL THEN isoform ELSE protein END as isoform,
COLLECT(type + ":" + CASE WHEN coordinate IS NOT NULL THEN coordinate ELSE "null" END) AS ptms
RETURN DISTINCT collect(DISTINCT pathway.stId), isoform, ptms
ORDER BY isoform, ptms
- Get number of reactions and pathways by each protein:
MATCH (p:Pathway{speciesName:"Homo sapiens"})-[:hasEvent*]->(r:Reaction{speciesName: "Homo sapiens"})-[:input|output|catalystActivity|physicalEntity|regulatedBy|regulator|hasComponent|hasMember|hasCandidate*]->(pe:PhysicalEntity{speciesName:'Homo sapiens'})-[:referenceEntity]->(re:ReferenceEntity{databaseName:'UniProt'})
RETURN DISTINCT count(DISTINCT p) as pathways, count(DISTINCT r) as reactions, re.identifier as protein
ORDER BY protein
- Get the number of hits by proteoform:
MATCH (p:Pathway{speciesName:"Homo sapiens"})-[:hasEvent*]->(r:Reaction{speciesName: "Homo sapiens"})-[:input|output|catalystActivity|physicalEntity|regulatedBy|regulator|hasComponent|hasMember|hasCandidate*]->(pe:PhysicalEntity{speciesName:'Homo sapiens'})-[:referenceEntity]->(re:ReferenceEntity{databaseName:'UniProt'})
WITH DISTINCT p, r, pe, re
OPTIONAL MATCH (pe)-[:hasModifiedResidue]->(tm:TranslationalModification)-[:psiMod]->(mod:PsiMod)
WITH DISTINCT p, r, pe.stId AS physicalEntity,
re.identifier AS protein,
re.variantIdentifier AS isoform,
tm.coordinate as coordinate,
mod.identifier as type ORDER BY type, coordinate
WITH DISTINCT p, r, physicalEntity,
protein,
CASE WHEN isoform IS NOT NULL THEN isoform ELSE protein END as isoform,
COLLECT(type + ":" + CASE WHEN coordinate IS NOT NULL THEN coordinate ELSE "null" END) AS ptms
RETURN DISTINCT count(DISTINCT p) as pathways, count(DISTINCT r) as reactions, (CASE WHEN isoform IS NOT NULL THEN isoform ELSE protein END + ptms) as proteoform
ORDER BY proteoform
- Get number of proteoforms per protein:
MATCH (pe:PhysicalEntity{speciesName:'Homo sapiens'})-[:referenceEntity]->(re:ReferenceEntity{databaseName:'UniProt'})
WITH DISTINCT pe, re
OPTIONAL MATCH (pe)-[:hasModifiedResidue]->(tm:TranslationalModification)-[:psiMod]->(mod:PsiMod)
WITH DISTINCT pe.stId AS physicalEntity,
re.identifier AS protein,
re.variantIdentifier AS isoform,
tm.coordinate as coordinate,
mod.identifier as type ORDER BY type, coordinate
WITH DISTINCT physicalEntity,
protein,
CASE WHEN isoform IS NOT NULL THEN isoform ELSE protein END as isoform,
COLLECT(type + ":" + CASE WHEN coordinate IS NOT NULL THEN coordinate ELSE "null" END) AS ptms
WITH DISTINCT protein, (CASE WHEN isoform IS NOT NULL THEN isoform ELSE protein END + ptms) as proteoform
RETURN protein, count(DISTINCT proteoform) as proteoforms
- Get average number of proteoforms:
MATCH (pe:PhysicalEntity{speciesName:'Homo sapiens'})-[:referenceEntity]->(re:ReferenceEntity{databaseName:'UniProt'})
WITH DISTINCT pe, re
OPTIONAL MATCH (pe)-[:hasModifiedResidue]->(tm:TranslationalModification)-[:psiMod]->(mod:PsiMod)
WITH DISTINCT pe.stId AS physicalEntity,
re.identifier AS protein,
re.variantIdentifier AS isoform,
tm.coordinate as coordinate,
mod.identifier as type ORDER BY type, coordinate
WITH DISTINCT physicalEntity,
protein,
CASE WHEN isoform IS NOT NULL THEN isoform ELSE protein END as isoform,
COLLECT(type + ":" + CASE WHEN coordinate IS NOT NULL THEN coordinate ELSE "null" END) AS ptms
WITH DISTINCT protein, (CASE WHEN isoform IS NOT NULL THEN isoform ELSE protein END + ptms) as proteoform
WITH protein, count(DISTINCT proteoform) as proteoforms
RETURN avg(proteoforms) as average_proteoforms
- Get average number of proteoforms for modified proteins:
MATCH (pe:PhysicalEntity{speciesName:'Homo sapiens'})-[:referenceEntity]->(re:ReferenceEntity{databaseName:'UniProt'})
WITH DISTINCT pe, re
MATCH (pe)-[:hasModifiedResidue]->(tm:TranslationalModification)-[:psiMod]->(mod:PsiMod)
WITH DISTINCT pe.stId AS physicalEntity,
re.identifier AS protein,
re.variantIdentifier AS isoform,
tm.coordinate as coordinate,
mod.identifier as type ORDER BY type, coordinate
WITH DISTINCT physicalEntity,
protein,
CASE WHEN isoform IS NOT NULL THEN isoform ELSE protein END as isoform,
COLLECT(type + ":" + CASE WHEN coordinate IS NOT NULL THEN coordinate ELSE "null" END) AS ptms
WITH DISTINCT protein, (CASE WHEN isoform IS NOT NULL THEN isoform ELSE protein END + ptms) as proteoform
WITH protein, count(DISTINCT proteoform) as proteoforms
RETURN avg(proteoforms) as average_proteoforms
- Run the script located at: src/R/1_Degree/average.R
- Get the protein and proteoform lists from the graph database in NEO4J console.
Gene list:
MATCH (ewas:EntityWithAccessionedSequence{speciesName:'Homo sapiens'})-[:referenceEntity]->(re:ReferenceEntity{databaseName:'UniProt'})
WITH re.identifier as protein, re.geneName as genes
WHERE size(genes) > 0
UNWIND genes as GENE
RETURN DISTINCT GENE
Protein list:
MATCH (pe:PhysicalEntity{speciesName:"Homo sapiens"})-[:referenceEntity]->(re:ReferenceEntity{databaseName:"UniProt"})
RETURN DISTINCT re.identifier as PROTEIN
Proteoform list:
MATCH (pe:PhysicalEntity{speciesName:'Homo sapiens'})-[:referenceEntity]->(re:ReferenceEntity{databaseName:'UniProt'})
WITH DISTINCT pe, re
OPTIONAL MATCH (pe)-[:hasModifiedResidue]->(tm:TranslationalModification)-[:psiMod]->(mod:PsiMod)
WITH DISTINCT pe,
re.identifier AS PROTEIN,
CASE WHEN re.variantIdentifier IS NOT NULL THEN re.variantIdentifier ELSE re.identifier END AS ISOFORM,
tm.coordinate as COORDINATE,
mod.identifier as TYPE
ORDER BY TYPE, COORDINATE
WITH DISTINCT pe, PROTEIN, ISOFORM,
COLLECT(TYPE + ":" + CASE WHEN COORDINATE IS NOT NULL THEN COORDINATE ELSE "null" END) AS PTMS
RETURN DISTINCT ISOFORM, PTMS
Then convert the proteoform format from NEO4J to SIMPLE. Use PathwayMatcher class called ProteoformFormatConverter.
java -cp PathwayMatcher.jar matcher.tools.ProteoformFormatConverter Reactome/Proteoforms/ all_proteoforms_neo4j_v72.csv all_proteoforms_v72.csv
- Find out the gene, protein and proteoform members of each pathway. For this we execute PathwayMatcher and get the whole search result.
Genes:
java -jar PathwayMatcher.jar -t gene -i reactome/all_genes.csv -o reactome/all_genes/
Proteins:
java -jar PathwayMatcher.jar -t uniprot -i reactome/all_proteins.csv -o reactome/all_proteins/
Proteoforms:
java -jar PathwayMatcher.jar -t proteoform -i reactome/all_proteoforms.csv -o reactome/all_proteoforms/ -m strict
- Execute main C++ program to create the pathway sets and calculate overlaps:
g++ src/3_rule_out_gene_centric_overlap/rule_out_gene_centric_overlap.cpp src/main.cpp -O3 -o Debug/analysis.exe
From the report file ("reports/3_rule_out_gene_centric_overlap_analysis.txt") choose a pair of pathways to see the variation in overlap.
- Get members of both pathways at the different levels of granularity. The pathway names, isoforms and post translational modifications responsible for decomposing the gene level only overlap will show up.
Genes:
MATCH (pathway:Pathway{speciesName:"Homo sapiens"})-[:hasEvent*]->(rle:Reaction{speciesName:"Homo sapiens"}),
(rle)-[:input|output|catalystActivity|physicalEntity|regulatedBy|regulator|hasComponent|hasMember|hasCandidate*]->(pe:PhysicalEntity{speciesName:"Homo sapiens"})-[:referenceEntity]->(re:ReferenceEntity{databaseName:'UniProt'})
WITH pathway, rle, re.identifier as protein, re.geneName as genes
WHERE size(genes) > 0 AND pathway.stId IN ["R-HSA-110056", "R-HSA-6783783"]
UNWIND genes as gene
WITH DISTINCT pathway, gene, protein
RETURN DISTINCT collect(DISTINCT pathway.stId), gene
Proteins:
MATCH (pathway:Pathway{speciesName:"Homo sapiens"})-[:hasEvent*]->(rle:Reaction{speciesName:"Homo sapiens"}),
(rle)-[:input|output|catalystActivity|physicalEntity|regulatedBy|regulator|hasComponent|hasMember|hasCandidate*]->(pe:PhysicalEntity{speciesName:"Homo sapiens"})-[:referenceEntity]->(re:ReferenceEntity{databaseName:'UniProt'})
WHERE pathway.stId IN ["R-HSA-110056", "R-HSA-6783783"]
RETURN DISTINCT collect(DISTINCT pathway.stId), re.identifier
Proteoforms:
MATCH (pathway:Pathway{speciesName:"Homo sapiens"})-[:hasEvent*]->(rle:Reaction{speciesName:"Homo sapiens"}),
(rle)-[:input|output|catalystActivity|physicalEntity|regulatedBy|regulator|hasComponent|hasMember|hasCandidate*]->(pe:PhysicalEntity{speciesName:"Homo sapiens"})-[:referenceEntity]->(re:ReferenceEntity{databaseName:'UniProt'})
WHERE pathway.stId IN ["R-HSA-109703", "R-HSA-111447"]
WITH DISTINCT pathway, rle, pe, re
OPTIONAL MATCH (pe)-[:hasModifiedResidue]->(tm:TranslationalModification)-[:psiMod]->(mod:PsiMod)
WITH DISTINCT pathway, rle.stId as reaction, pe.stId AS physicalEntity,
re.identifier AS protein, re.variantIdentifier AS isoform, tm.coordinate as coordinate,
mod.identifier as type
ORDER BY type, coordinate
WITH DISTINCT pathway, reaction, physicalEntity, protein,
CASE WHEN isoform IS NOT NULL THEN isoform ELSE protein END as isoform,
COLLECT(type + ":" + CASE WHEN coordinate IS NOT NULL THEN coordinate ELSE "null" END) AS ptms
RETURN DISTINCT collect(DISTINCT pathway.stId), isoform, ptms
ORDER BY isoform, ptms
Follow the same steps of the gene level only overlap.
- Download the data of all GWAS from https://www.ncbi.nlm.nih.gov/gap/phegeni and store it at resources/PheGenI/
Obtain the number of reactions and pathways where each protein or proteoform participates in the Reactome database:
- Number of reactions and pathways for each protein:
MATCH (p:Pathway{speciesName:"Homo sapiens"})-[:hasEvent*]->
(r:Reaction{speciesName: "Homo sapiens"})-[:input|output|catalystActivity|physicalEntity|regulatedBy|regulator|hasComponent|hasMember|hasCandidate*]->
(pe:PhysicalEntity{speciesName:'Homo sapiens'})-[:referenceEntity]->(re:ReferenceEntity{databaseName:'UniProt'})
WITH DISTINCT p, r, re.identifier as PROTEIN
RETURN DISTINCT PROTEIN, count(r) as NUM_REACTIONS, count(p) as NUM_PATHWAYS
ORDER BY PROTEIN
Store the result data to a file called: "num_pathways_per_protein.csv"
- Number of reactions and pathways for each proteoform:
MATCH (p:Pathway{speciesName:"Homo sapiens"})-[:hasEvent*]->
(r:Reaction{speciesName: "Homo sapiens"})-[:input|output|catalystActivity|physicalEntity|regulatedBy|regulator|hasComponent|hasMember|hasCandidate*]->
(pe:PhysicalEntity{speciesName:'Homo sapiens'})-[:referenceEntity]->(re:ReferenceEntity{databaseName:'UniProt'})
WITH DISTINCT p, r, pe, re
OPTIONAL MATCH (pe)-[:hasModifiedResidue]->(tm:TranslationalModification)-[:psiMod]->(mod:PsiMod)
WITH DISTINCT p, r, pe,
re.identifier AS PROTEIN,
CASE WHEN re.variantIdentifier IS NOT NULL THEN re.variantIdentifier ELSE re.identifier END AS ISOFORM,
tm.coordinate as COORDINATE,
mod.identifier as TYPE
ORDER BY TYPE, COORDINATE
WITH DISTINCT p, r, pe, PROTEIN, ISOFORM,
COLLECT(TYPE + ":" + CASE WHEN COORDINATE IS NOT NULL THEN COORDINATE ELSE "null" END) AS PTMS
WITH DISTINCT p, r, pe, PROTEIN, ISOFORM + PTMS as PROTEOFORM
RETURN DISTINCT PROTEIN, PROTEOFORM, count(r) as NUM_REACTIONS, count(p) as NUM_PATHWAYS
ORDER BY PROTEIN, PROTEOFORM
Store the result data to a file called: "num_pathways_per_proteoform.csv"
Note: These two previous queries only show the proteins and proteoforms that have an annotation stating that they participate in at least a Reaction and a Pathway.
- Get reactions and pathways where each gene participates:
MATCH (p:Pathway{speciesName:"Homo sapiens"})-[:hasEvent*]->(r:Reaction{speciesName: "Homo sapiens"})-[:input|output|catalystActivity|physicalEntity|regulatedBy|regulator|hasComponent|hasMember|hasCandidate*]->(pe:PhysicalEntity{speciesName:'Homo sapiens'})-[:referenceEntity]->(re:ReferenceEntity{databaseName:'UniProt'})
WHERE size(re.geneName) > 0
UNWIND re.geneName as GENE
RETURN DISTINCT GENE, r.stId as REACTION_STID, p.stId as PATHWAY_STID, re.identifier as PROTEIN
ORDER BY GENE, PROTEIN, PATHWAY_STID, REACTION_STID
- Get reactions and pathways where each protein participates:
MATCH (p:Pathway{speciesName:"Homo sapiens"})-[:hasEvent*]->(r:Reaction{speciesName:"Homo sapiens"}),
(r)-[:input|output|catalystActivity|physicalEntity|regulatedBy|regulator|hasComponent|hasMember|hasCandidate*]->(pe:PhysicalEntity{speciesName:"Homo sapiens"}),
(pe)-[:referenceEntity]->(re:ReferenceEntity{databaseName:"UniProt"})
RETURN DISTINCT re.identifier as PROTEIN, r.stId as REACTION_STID, p.stId as PATHWAY_STID, r.displayName as REACTION_NAME, p.displayName as PATHWAY_NAME
ORDER BY PROTEIN, PATHWAY_STID, REACTION_STID
- Get reactions and pathways where each proteoform participates:
MATCH (p:Pathway{speciesName:"Homo sapiens"})-[:hasEvent*]->(r:Reaction{speciesName: "Homo sapiens"})-[:input|output|catalystActivity|physicalEntity|regulatedBy|regulator|hasComponent|hasMember|hasCandidate*]->(pe:PhysicalEntity{speciesName:'Homo sapiens'})-[:referenceEntity]->(re:ReferenceEntity{databaseName:'UniProt'})
WITH DISTINCT p, r, pe, re
OPTIONAL MATCH (pe)-[:hasModifiedResidue]->(tm:TranslationalModification)-[:psiMod]->(mod:PsiMod)
WITH DISTINCT p, r, pe,
re.identifier AS PROTEIN,
CASE WHEN re.variantIdentifier IS NOT NULL THEN re.variantIdentifier ELSE re.identifier END AS ISOFORM,
tm.coordinate as COORDINATE,
mod.identifier as TYPE
ORDER BY TYPE, COORDINATE
WITH DISTINCT p, r, pe, PROTEIN, ISOFORM, COLLECT(TYPE + ":" + CASE WHEN COORDINATE IS NOT NULL THEN COORDINATE ELSE "null" END) AS PTMS
WITH DISTINCT p, r, pe, PROTEIN, ISOFORM + ";" + PTMS as PROTEOFORM
RETURN DISTINCT PROTEOFORM, r.stId as REACTION_STID, p.stId as PATHWAY_STID
ORDER BY PROTEOFORM, PATHWAY_STID, REACTION_STID
[1] McLaren, W. et al. The Ensembl Variant Effect Predictor. Genome Biology 17, 122, doi:10.1186/s13059-016-0974-4 (2016).
[2] Ramos, Erin M., et al. "Phenotype–Genotype Integrator (PheGenI): synthesizing genome-wide association study (GWAS) data with existing genomic resources." European Journal of Human Genetics 22.1 (2014): 144.
[3] Menche, Jörg, et al. "Uncovering disease-disease relationships through the incomplete interactome." Science 347.6224 (2015): 1257601.
[4] Fabregat, A. et al. The Reactome Pathway Knowledgebase. Nucleic acids research 46, D649-d655, doi:10.1093/nar/gkx1132 (2018).
[5] Ramos, Erin M., et al. "Phenotype–Genotype Integrator (PheGenI): synthesizing genome-wide association study (GWAS) data with existing genomic resources." European Journal of Human Genetics 22.1 (2014): 144.
[6] Menche, Jörg, et al. "Uncovering disease-disease relationships through the incomplete interactome." Science 347.6224 (2015): 1257601.