QC and data validation with lcmsWorld - PGB-LIV/lcmsWorld GitHub Wiki
lcmsWorld can be used for a variety of data validation and quality-control checks. While this list is not exhaustive, it gives some examples of how lcmsWorld can be used to help check that the LC-MS data, and identications, have been processed correctly.
Confirming the effectiveness of labelling
Given a view of the data, it is easy to see frequent pairs of isotope patterns, confirming that there was a reasonable uptake of SILAC labelling, such as in Figure 1.
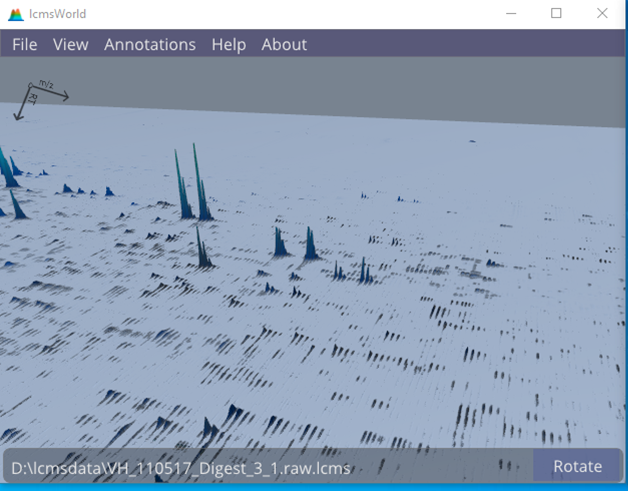 Figure 1. View of SILAC labelled file, showing many matching pairs of peaks.
In a closer inspection, the distance between these pairs of peaks can be measured by holding the cursor over each, and viewing the reported m/z values. In Figure 2, we can see that the mass difference is 6.003 da (554.309 – 548.306), consistent with heavy arginine SILAC labelling.
Figure 1. View of SILAC labelled file, showing many matching pairs of peaks.
In a closer inspection, the distance between these pairs of peaks can be measured by holding the cursor over each, and viewing the reported m/z values. In Figure 2, we can see that the mass difference is 6.003 da (554.309 – 548.306), consistent with heavy arginine SILAC labelling.
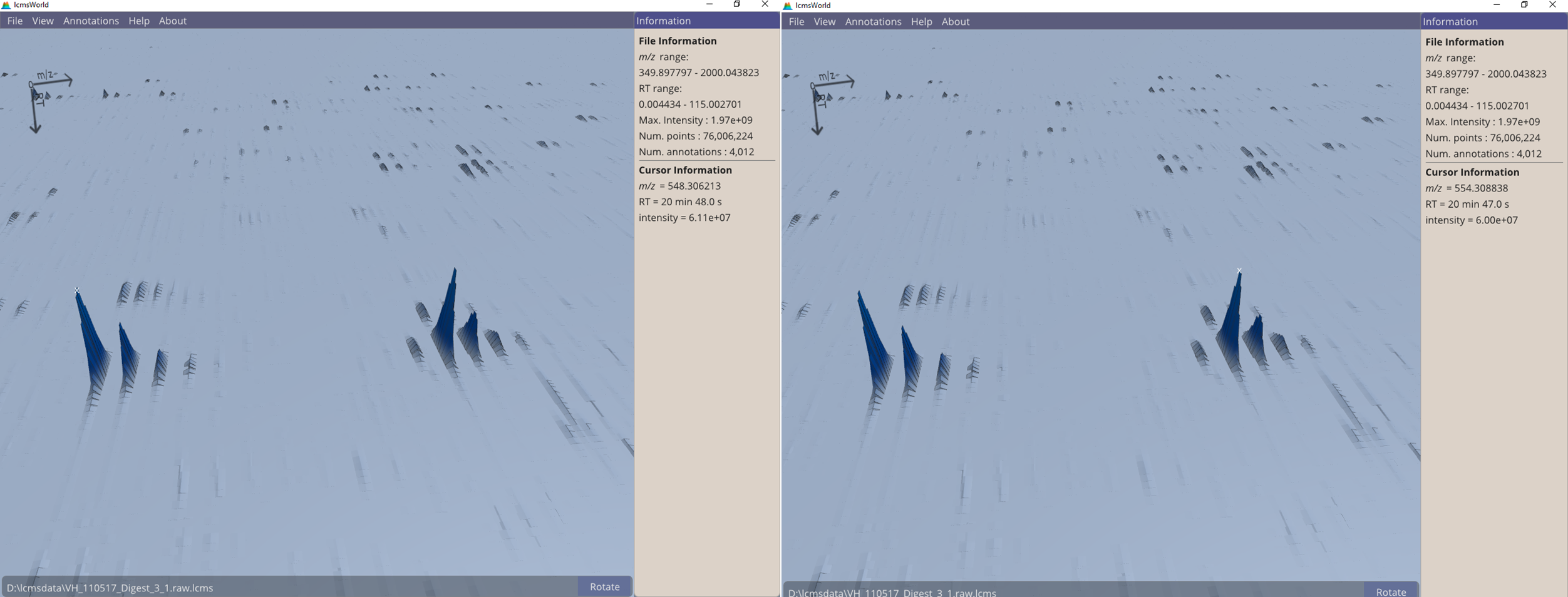 Figure 2. View of SILAC labelled file, showing the measurement of two matching peaks.
Figure 2. View of SILAC labelled file, showing the measurement of two matching peaks.
Confirming identification data matches
To confirm if the identification data is actually derived from the original raw data file, it is possible to load the identifications alongside the raw data, and then quickly check that identifications match to the peaks in the data. All identifications can be highlighted (from annotations menu, ‘mark annotations’), which gives a quick impression of how much of the data was related to an identification, shown in Figure 3.
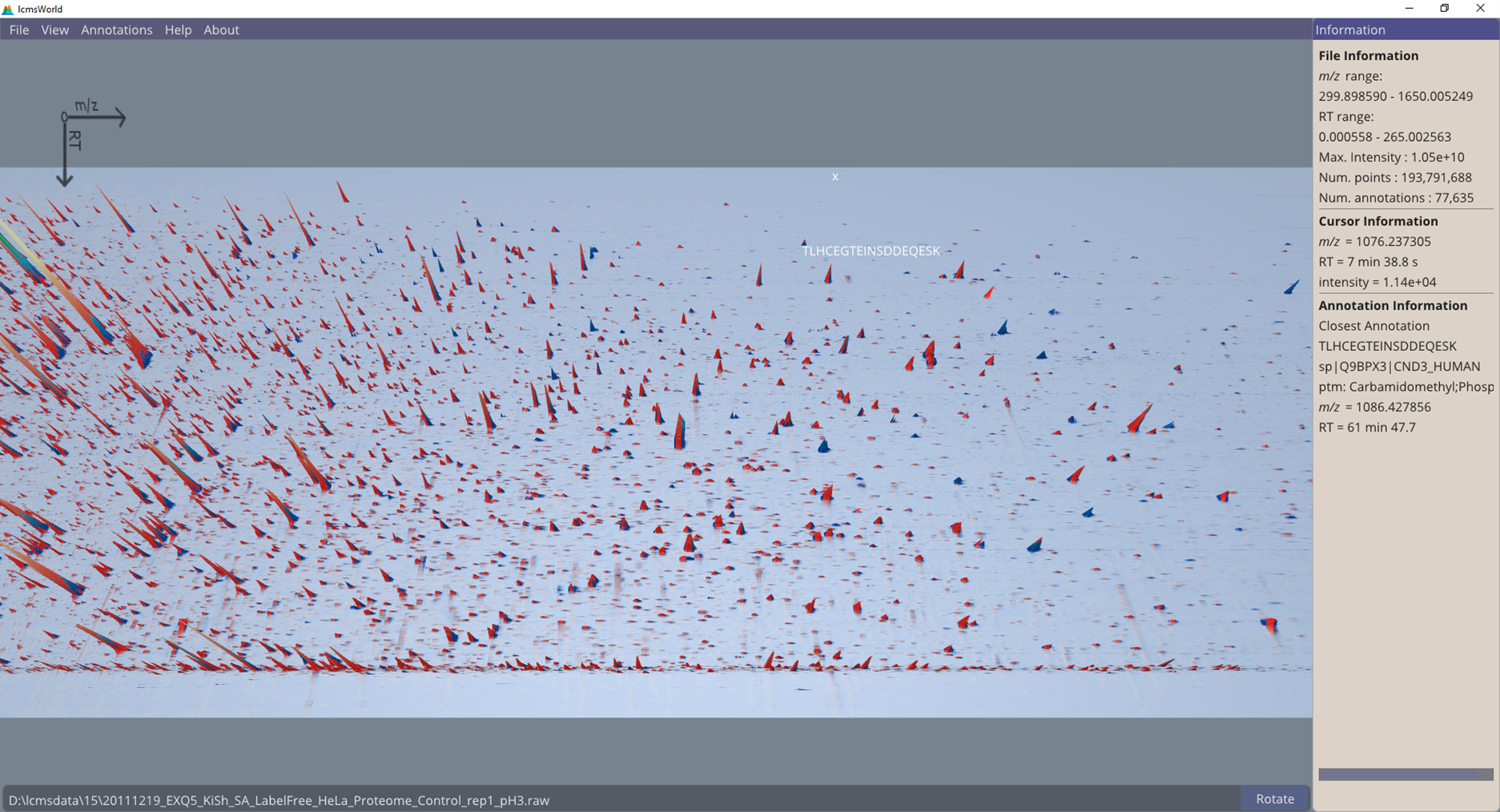 Figure 3. Data view highlighting regions with identified peptides.
Multiple annotations can also be highlighted, which gives an impression of how well the identifications match to the peaks in the data, Figure 4. Where these identifications do not match peaks in the data could be an indication that is a mismatch between the identification file and the raw data file.
Figure 3. Data view highlighting regions with identified peptides.
Multiple annotations can also be highlighted, which gives an impression of how well the identifications match to the peaks in the data, Figure 4. Where these identifications do not match peaks in the data could be an indication that is a mismatch between the identification file and the raw data file.
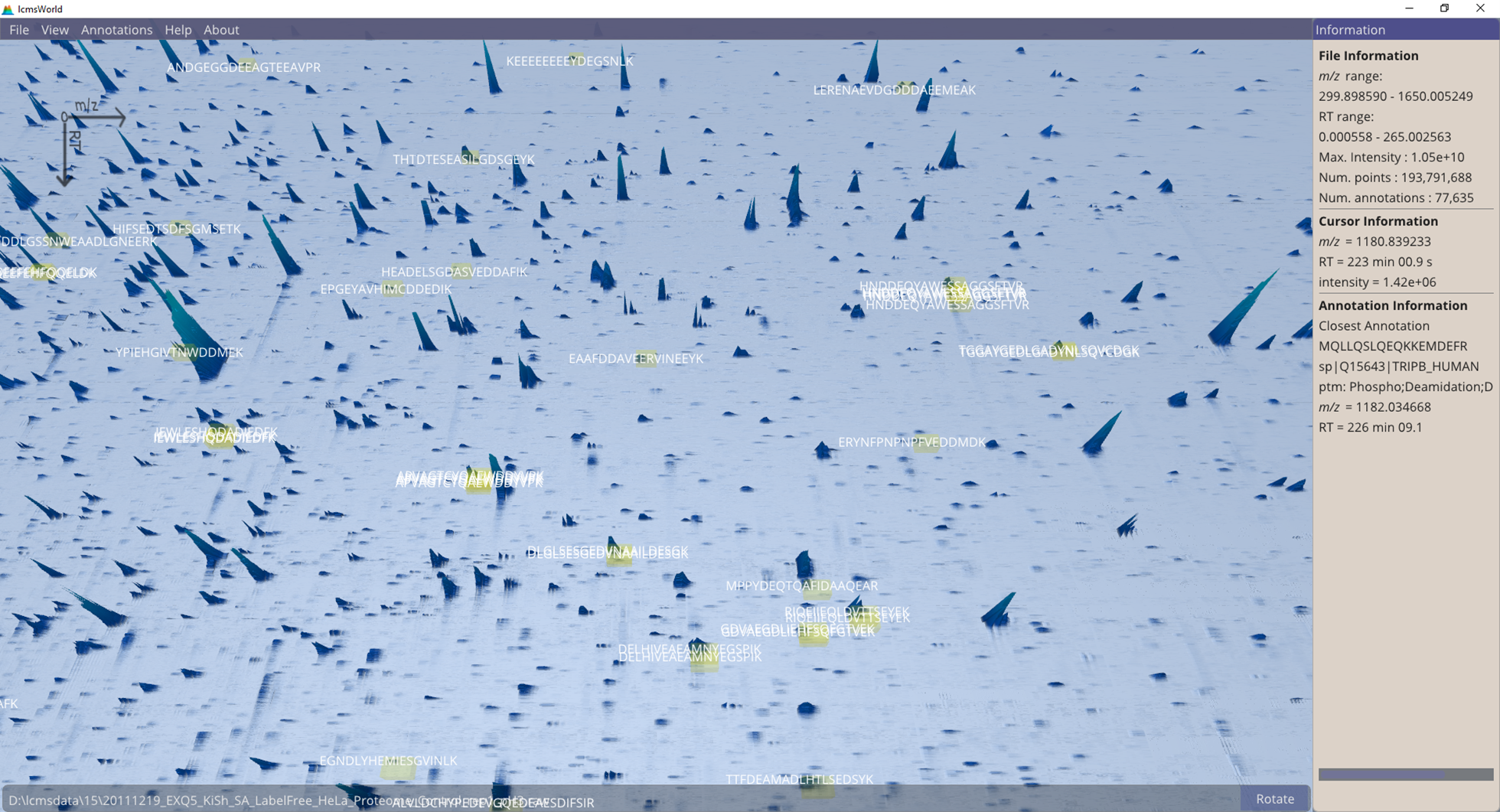 Figure 4. A closer view of the data with multiple individual identifications highlighted.
Figure 4. A closer view of the data with multiple individual identifications highlighted.
Individual identifications can also be selected, either from a menu or by navigating around the data view, to check that the corresponding data looks like a plausible isotope pattern, as in Figure 5.
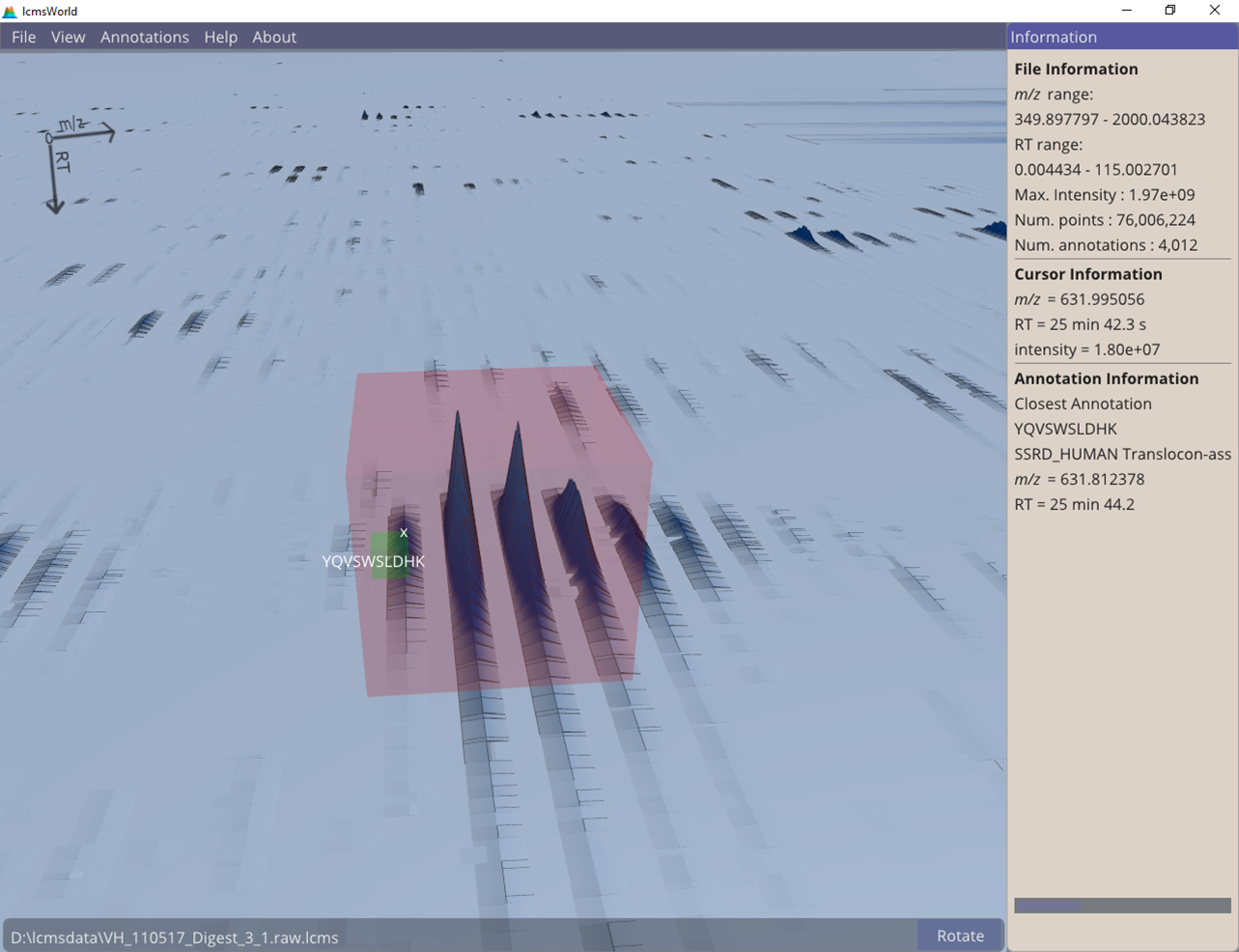 Figure 5. Data view of a single identified peptide.
Figure 5. Data view of a single identified peptide.
Verifying replication
Simultaneously viewing two or more different data files makes it easy to get a quick impression of the quality of any replication. In the example shown in Figure 6, it is quickly obvious that there is good agreement between the different replications.
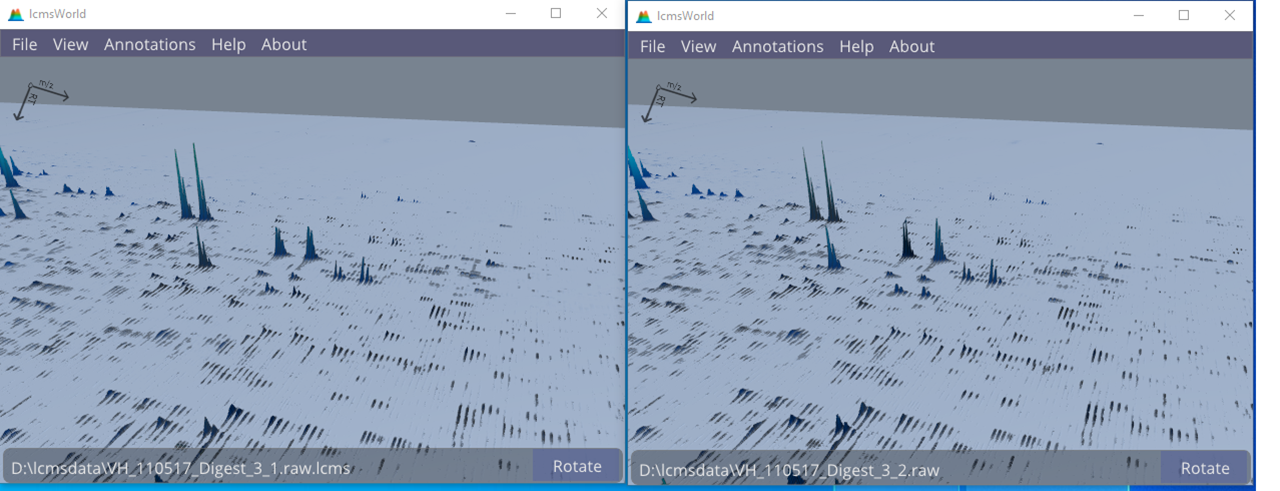 Figure 6. Comparing the data from two replicates, showing many similarities.
By viewing the data in high detail, as in Figure 7, it is also possible to verify that these are actually replicates and not, for example, caused by a duplicated data file.
Figure 6. Comparing the data from two replicates, showing many similarities.
By viewing the data in high detail, as in Figure 7, it is also possible to verify that these are actually replicates and not, for example, caused by a duplicated data file.
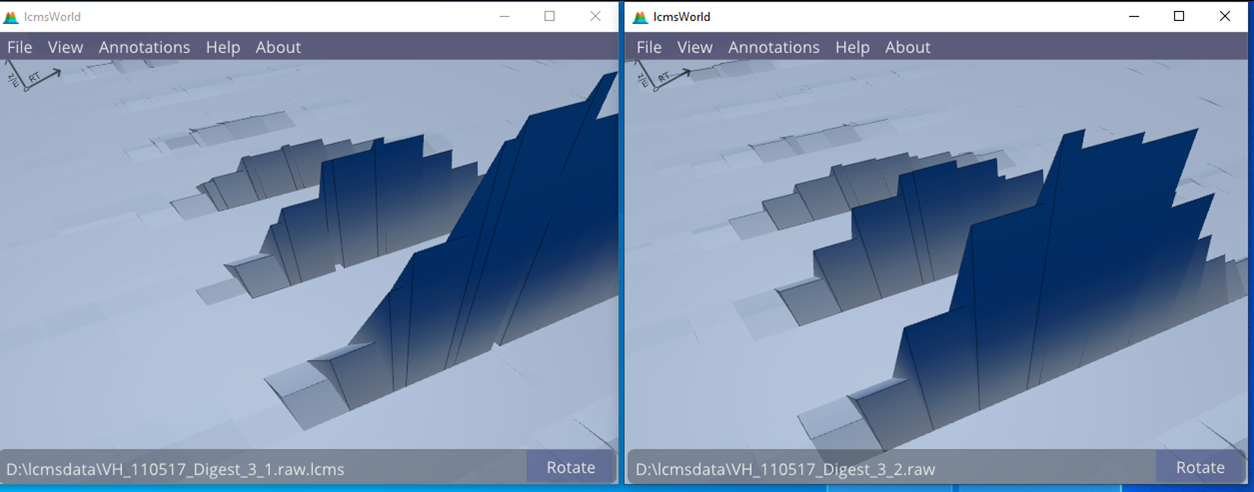 Figure 7. Comparing the data from two replicates, showing slight differences in the data.
Figure 7. Comparing the data from two replicates, showing slight differences in the data.
Checking identifications and contamination
By comparing identifications that are made across multiple files, it is possible to compare the data. In this example, Figure 8, we find that 3 of 4 species contain the same identification. In the 4th species (top-right), there is clearly a matching data peak, but no identification was made. This was possibly due to being conflated with the data from another nearby, but larger, peak.
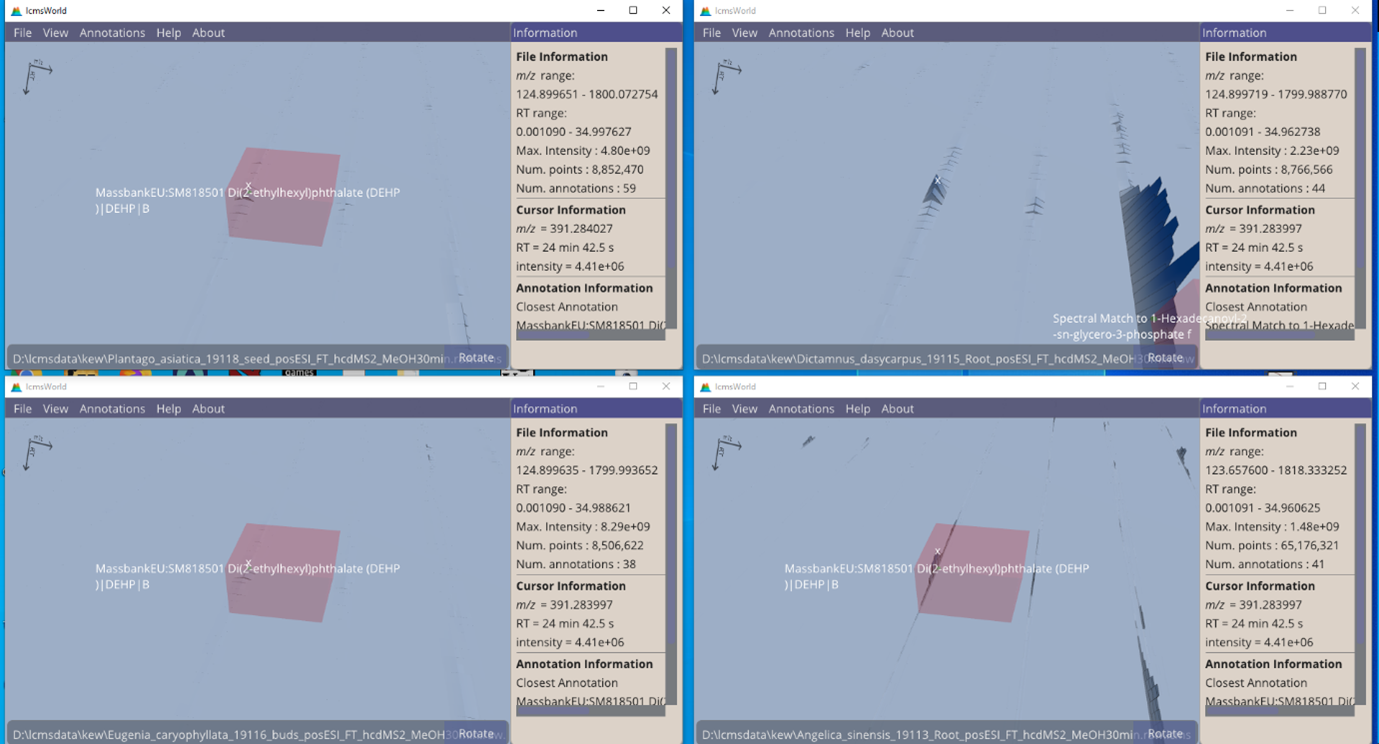 Figure 8. Four data files viewed simultaneously, with 3 identifications highlighted.
Figure 8. Four data files viewed simultaneously, with 3 identifications highlighted.
Obviously care should be taken when relying on identifications from other species; in the example of Figure 9 there is a matching data peak in a different species. In this case, it was found to be appearing as a result of contamination from the first sample due to an over-running LC column.
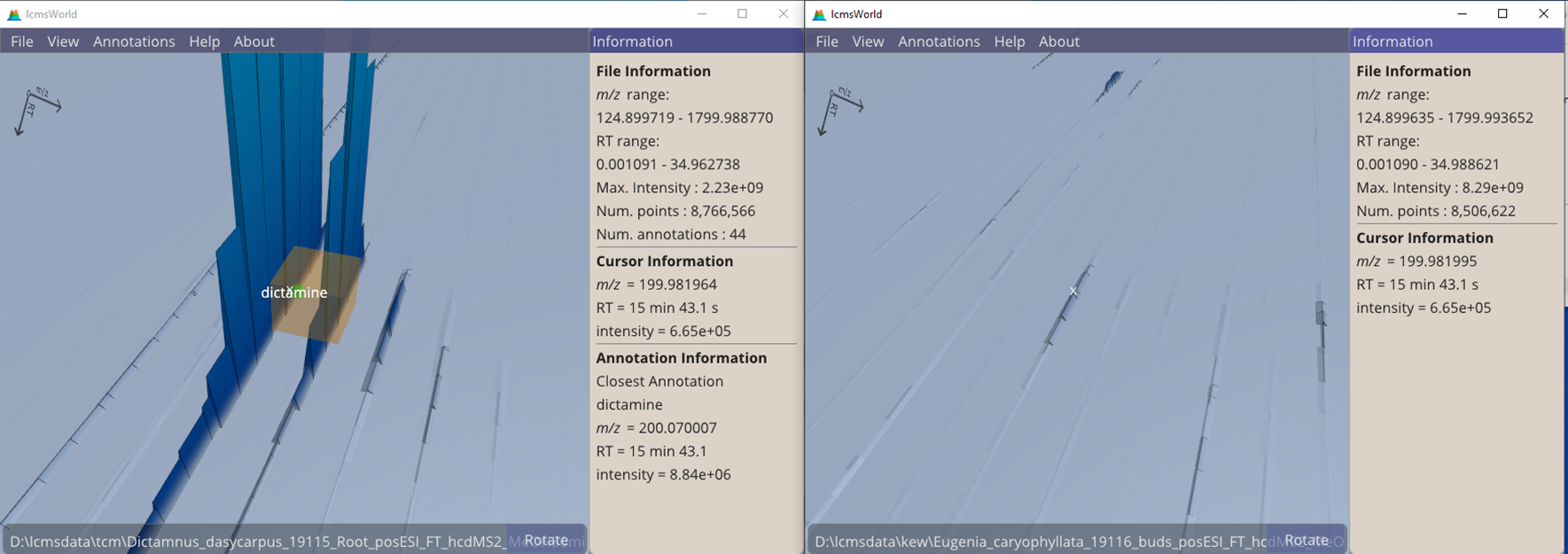 Figure 9. Two data files showing a small peak appearing in the second file as a result of contamination.
It is also easy to check the data where an identification may have been expected, but was not found. In this example, Figure 10, we have clear evidence of the presence of some peptide, but no feature was found by the software. It appears that there are multiple overlapping features that would make automatic identification more difficult.
Figure 9. Two data files showing a small peak appearing in the second file as a result of contamination.
It is also easy to check the data where an identification may have been expected, but was not found. In this example, Figure 10, we have clear evidence of the presence of some peptide, but no feature was found by the software. It appears that there are multiple overlapping features that would make automatic identification more difficult.
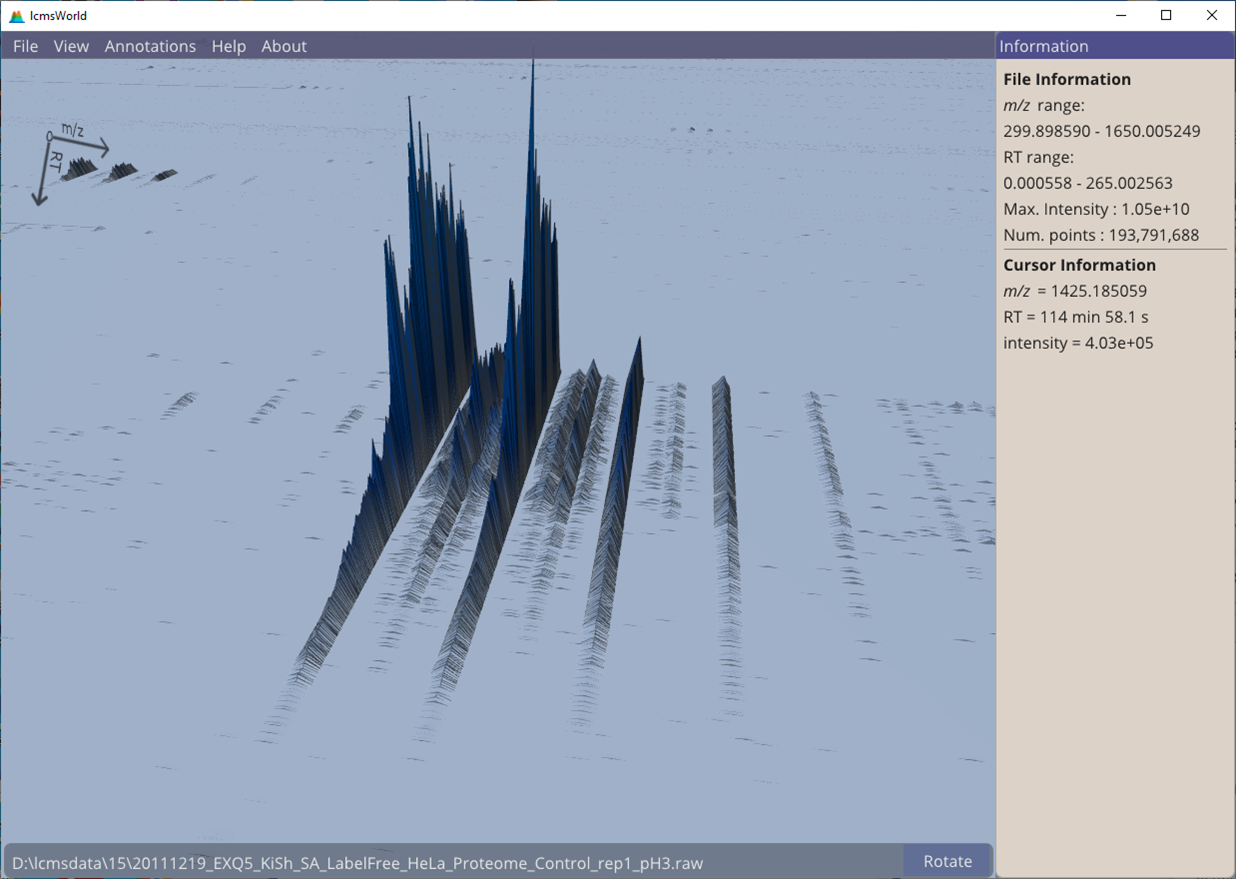
Figure 10. Clear isotope patterns in the data, but where no identification was made.