Metagenomics Tutorial Answers - LangilleLab/microbiome_helper GitHub Wiki
These are the answers to the questions from the Metagenomics Tutorial.
- 12 - since these are paired-end reads there is a forward and reverse FASTQ for each sample.
- 6 - from the mapping file there are 6 Cancer and 6 Normal samples.
- Each FASTQ is 300,000 lines long, which means there are 75,000 paired-end reads in each one.
- 1832 reads
- humann2 v0.11.1 and MetaPhlAn2 version 2.5.0 (28 April 2015) were installed when this tutorial was written. Keeping track of this information (including for the other programs in your pipeline) is important for reproducibility.
- 17.8%
- 14.8%
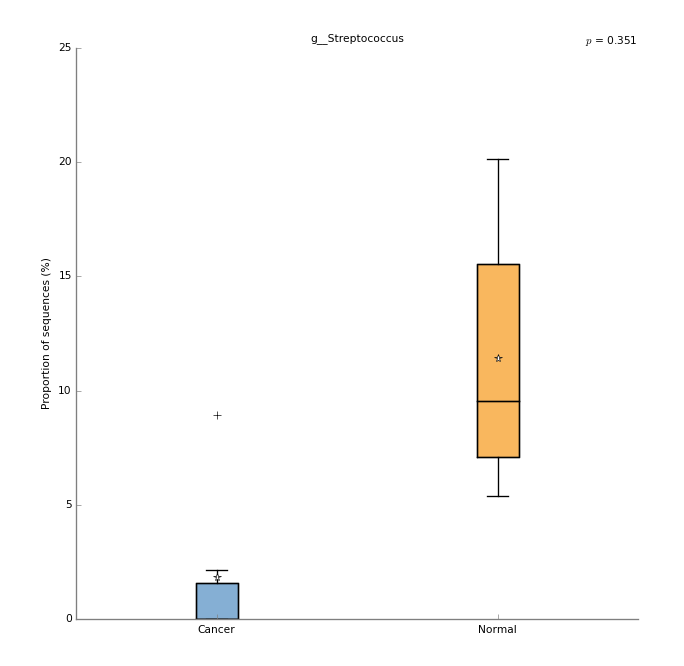
- g__Streptococcus
- You should have saved a plot like this: images/mgs_tutorial_2017_images/g__Streptococcus_boxplot.png
- P=0.004
{kind=link}