CBW 2018 Metagenomic Taxonomic and Functional Composition Tutorial Answers - LangilleLab/microbiome_helper GitHub Wiki
These are the answers for the Metagenomic Taxonomic and Functional Composition Tutorial created for the 2018 microbiome data analysis Canadian Bioinformatics Workshop.
-
Yes, there should be the same number of reads in the reverse FASTQ for each sample. So there should be 100000/4 = 25000 for sample CSM79HR8 and 100400/4 = 25100 for sample HSM7J4QT.
-
Only 4 reads were removed due to matching the human and/or PhiX genomes, which again highlights that this data has already been stringently filtered.
-
Researchers have different preferences and opinions about how raw data should be pre-processed. It's important to upload all the raw data so that your work can be fully reproducible and so different pipelines could be used.
-
The configured nucleotide database is
/usr/local/lib/python2.7/dist-packages/humann2/data/chocophlan_DEMOand the protein database is/usr/local/lib/python2.7/dist-packages/humann2/data/uniref_DEMO. -
The pathway-level inferences should be the same! This is because the E.C. numbers are used to infer the pathway levels - UniRef gene families that are cannot be linked to individual reactions are not used.
-
If both the forward and reverse reads match the same gene they will count as 2 independent hits! In practice, this shouldn't matter since shotgun metagenomics sequencing sample depths tend to be so high. However, this could be important to think about if you think the average gene sizes across your samples could differ.
-
The UniRef90_unknown category corresponds to reads mapped at the nucleotide alignment step that mapped to reads without UniRef90 ids. In contrast, reads in the UNMAPPED category didn't map to anything!
-
No you can't since the reported values are reads per kilo-base (RPK); you would need to know the size of each gene family to figure out the number of mapped reads from this table.
-
No, the pathway abundances reported for each individual taxa do not sum to the community-wide abundance. This is because the community-level pathway abundance is based on the abundance of gene families across all taxa. This becomes easier to understand if you imagine gene family X that is abundant in organism Y and is required for a pathway, but no other gene families required for that pathway are present in organism Y. In this case organism Y wouldn't show any evidence of producing this pathway, but the abundance of gene family X in organism Y would contribute to the community-wide abundance of the pathway!
-
Yes, for instance:
PWY-7220: adenosine deoxyribonucleotides de novo biosynthesis II. We have no evidence based on this output file that this pathway is within an individual species - the required genes might just be present across multiple taxa! However, it's also possible that species containing all the requisite enzymes for this pathway simply aren't in the ChocoPhlAn database. -
62.75%
-
There are 5 independent species identified. You could figure that out a number of ways, but one way would be to type this command:
grep -v t__ humann2_out/HSM7J4QT/HSM7J4QT_humann2_temp/HSM7J4QT_metaphlan_bugs_list.tsv | grep s__ -
There are 232 species.
-
Since MetaPhlAn2 is marker-based approach it is important that the exact clade-specific marker gene is matched to the reads. If the
--localoption is used then reads would be allowed to only partially align to the marker genes, which would mean that reads matching homologs in different taxa (and random matches) would increase the false positive rate. -
9 Bacteroides species were identified by
centrifugeand 2 by MetaPhlAn2. -
Dialister invisus has an abundance of 10.26% based on MetaPhlAn2, but wasn't found at all by
centrifuge! -
This command would run MetaPhlAn2 on all samples in that directory with
parallel:parallel --eta -j 4 'metaphlan2.py {} example_strainphlan2_out/{/.}_profile.txt \ --bowtie2out example_strainphlan2_out/{/.}_bowtie2.txt \ --samout example_strainphlan2_out/{/.}.sam.bz2 \ --input_type fastq' ::: cat_reads/*fastq -
You need to run
class(bovatus_tree)andclass(bovatus_gg)to answer this question.bovatus_treeis a "phylo" object which is widely used tree class used by several R packages. In contrast,bovatus_ggis a more specialized object that returns these 3 classes: "ggtree" "gg" "ggplot". -
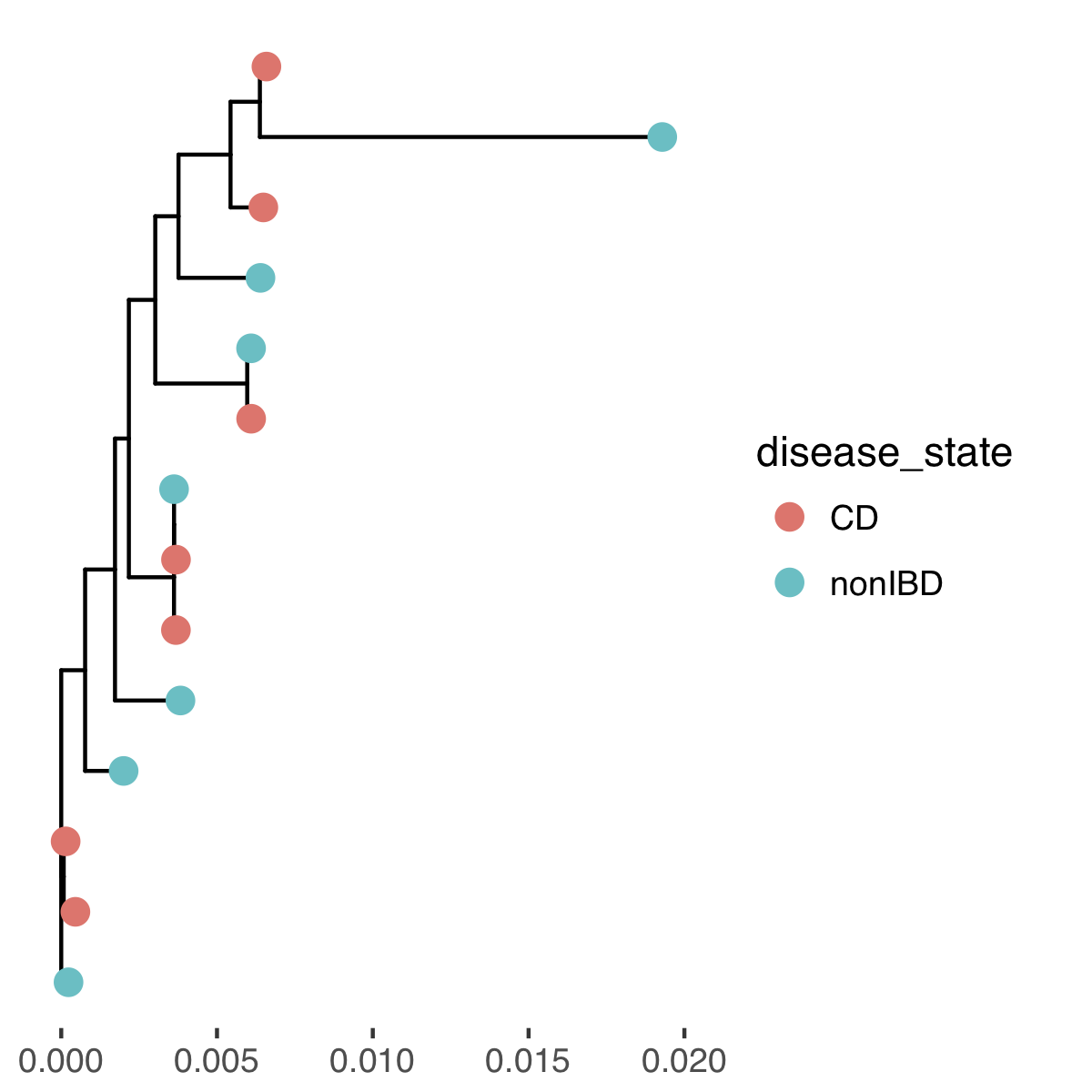
Your plotted tree should look like this:
images/CBW_2018/b_ovatus_ggtree.png
{kind=link}
Since the Crohn's disease and control patients are not clustering separately we do not have support for our hypothesis!