2. Method - Kkkzq/Genome-Analysis-paper2 GitHub Wiki
FastQC (Andrews S., 2010) is used to check the quality of reads. It requires data from BAM, SAM, or FastQ files and provides summary tables and graphs to evaluate the quality of data. It works with the following code format (See scripts here):
fastqc filename.fastq.gz -o output/directory
See result details here.
Trimmomatic (Bolger et al., 2014) can trim Illumina(FASTQ) data and remove adapters. The code format for single end mode is:
java -classpath <path to trimmomatic jar> org.usadellab.trimmomatic.TrimmomaticSE [-threads <threads>] [-phred33 | -phred64] [-trimlog <logFile>] <input> <output> <step 1> ...
The format for paired end mode is:
java -classpath <path to trimmomatic jar> org.usadellab.trimmomatic.TrimmomaticPE [-
threads <threads>] [-phred33 | -phred64] [-trimlog <logFile>] [-basein <inputBase> | <input
1> <input 2>] [-baseout <outputBase> | <paired output 1> <unpaired output 1> <paired
output 2> <unpaired output 2> <step 1> ...
- 2 input files: forward & reverse
- 4 output files
- mySampleFiltered_1P.fq.gz - for paired forward reads
- mySampleFiltered_1U.fq.gz - for unpaired forward reads
- mySampleFiltered_2P.fq.gz - for paired reverse reads
- mySampleFiltered_2U.fq.gz - for unpaired reverse reads
The parameters for Trimmoatic are specified as following codes. The full script could be seen here.
ILLUMINACLIP:/sw/bioinfo/trimmomatic/0.36/rackham/adapters/TruSeq3-PE-2.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36
- ILLUMINACLIP: cuts adapters and other Illumina specific sequences;
- PE: specifies the data is paired-end;
- LEADING: cuts the bases off the start of the read if they are below 3;
- TRAILING: cuts the bases off the end of the read if they are below 3;
- SLIDINGWINDOW: scan the read with a 4 base window and cut when the average quality per base falls below 15;
- MINLEN: drops the read if it's smaller than 36bp after the previous steps have been done;
SOAPdenovo(Luo et al., 2012) and SPAdes (Nurk et al., 2013) are used to finish genome assembly.
SOAPdenoco was designed for Illumina GA short reads.
1. Configuration file
A configuration file is needed to specify the path to files in FASTA, FASTQ, BAM format, and relevant information by the following items.
# Reads ..96
[LIB]
avg_ins=9000
reverse_seq=1
asm_flags=2
rank=1
rd_len_cutoff=100
pair_num_cutoff=5
map_len=35
q1=/home/ziqi/Ziqi_Genome_Analysis/2_Eckalbar_2016/sel3/wgs_data/sel3_SRR5819796.trim_1P.fastq.gz
q2=/home/ziqi/Ziqi_Genome_Analysis/2_Eckalbar_2016/sel3/wgs_data/sel3_SRR5819796.trim_2P.fastq.gz
q=/home/ziqi/Ziqi_Genome_Analysis/2_Eckalbar_2016/sel3/wgs_data/sel3_SRR5819796.trim_1U.fastq.gz
q=/home/ziqi/Ziqi_Genome_Analysis/2_Eckalbar_2016/sel3/wgs_data/sel3_SRR5819796.trim_2U.fastq.gz
-
avg_ins: the average insert size of this library.
-
reverse_seq: Should be value 0 or 1. It tells the assembler if the read sequences need to be complementarily reversed (0, forward-reverse; 1, reverse-forward).
-
asm_flags: This indicator decides in which part(s) the reads are used. It takes value 1(only contig assembly), 2 (only scaffold assembly), 3(both contig and scaffold assembly), or 4 (only gap closure).
-
rd_len_cutof: The assembler will cut the reads from the current library to this length.
-
rank: It takes integer values and decides in which order the reads are used for scaffold assembly.
-
pair_num_cutoff: This parameter is the cutoff value of pair numbers for a reliable connection between two contigs or pre-scaffolds. The minimum number for paired-end reads and mate-pair reads is 3 and 5 respectively.
-
map_len: The minimum alignment length between a read and a contig required for a reliable read location. The minimum length for paired-end reads and mate-pair reads is 32 and 35 respectively.
-
q1 and q2: paired reads in two fastq sequence files;
-
q: single end fasta files;
2. Code format
${bin} all -s config_file -K 63 -R -o graph_prefix 1>ass.log 2>ass.err
- -s: specify the config file
- -K: Kmer size. Choose 49 because of the paper
- -o: specify the output prefix
See full scripts here.
3. Output files
- .contig: contig sequences without using mate-pair information.
- .scafSeq: scaffold sequences (final contig sequences can be extracted by breaking down scaffold sequences at gap regions
GapCloser is used to improve the assembly SOAPdenovo result files.
GapCloser -b config.txt -a out.scafSeq -o gapcloser.fasta
- -b: the path of the config file used during the SOAPdenovo assembly
- -a: the .scafSeq resulting from SOAPdenovo
- -o: the output file name
The output fasta file contains the scaffold sequences with gaps filled. Another *.fill file describes the information of the gaps in the scaffolds.
SPAdes supports paired-end reads, mate-pairs, and unpaired reads. It is worth mentioning that SPAdes was initially designed for small genomes, thus it might not perform well in larger genomes.
For command line format:
spades.py [options] -o <output_dir>
See full script here.
-
-o: specify the output directory
-
--pe<#>-1 <file_name>: Input file with left reads for paired-end library number <#> (<#> = 1,2,..,9).
-
--pe<#>-2 <file_name>: Input file with right reads for paired-end library number <#> (<#> = 1,2,..,9).
-
--pe<#>-s <file_name>: File with unpaired reads from paired-end library number <#> (<#> = 1,2,..,9)
-
--mp<#>-1 <file_name>: Input file with left reads for mate-pair library number <#> (<#> = 1,2,..,9).
-
--mp<#>-2 <file_name>: Input file with right reads for mate-pair library number <#> (<#> = 1,2,..,9).
For the output files:
- scaffolds.fasta contains resulting scaffolds (recommended for use as resulting sequences)
- contigs.fasta contains resulting contigs
MUMmer is an open-source software package for the rapid alignment of very large DNA and amino acid sequences. In this project, MUMmer is used to evaluating the assemblies. NUCmer (NUCleotide MUMmer) is the most user-friendly alignment script for standard DNA sequence alignment. It is a robust pipeline that allows for multiple reference and multiple query sequences to be aligned in a many vs. many fashion. See full scripts here.
A code example:
nucmer --prefix=nucmer reference.fasta your_assembly.fasta # outputs a *.delta file, needs to be visualized
mummerplot -x "[0,4000000]" -y "[0,4000000]" --png --layout --filter -p mummer nucmer.delta -R reference.fasta -Q your_assembly.fasta
Mummerplot can output several files, but only the *.png image was used in my evaluation. The mummerplot script outputs three files, .gp .fplot .rplot, when run with standard parameters. The first of which is the gnuplot script. This script contains the commands necessary to generate the plot, and refers to the two data files which contain the forward and reverse matches respectively. If the --filter or --layout option are specified, an additional .filter file will be generated containing the filtered delta information. The plot graphic will be saved to the file .ps or .png if specified.
See the details about the result figures in the result part.
Trinity assembles transcript sequences from Illumina RNA-Seq data. Trinity combines three independent software modules: Inchworm, Chrysalis, and Butterfly, applied sequentially to process large volumes of RNA-seq reads.
Trinity partitions the sequence data into many individual de Bruijn graphs, each representing the transcriptional complexity at a given gene or locus, and then processes each graph independently to extract full-length splicing isoforms and to tease apart transcripts derived from paralogous genes.
A typical trinity code will be like this:
Trinity --seqType fq --max_memory 50G \
--left reads_1.fq.gz --right reads_2.fq.gz --CPU 6
# If there are multiple pairs of fastq files
Trinity --seqType fq --max_memory 50G \
--left condA_1.fq.gz,condB_1.fq.gz,condC_1.fq.gz \
--right condA_2.fq.gz,condB_2.fq.gz,condC_2.fq.gz \
--CPU 6
In order to use RepeatMasker, it is necessary to compile a specific repeat library using RepeatScout.
mkdir -p masking
build_lmer_table -sequence data/genome.fa -freq masking/genome.freq
RepeatScout -sequence data/genome.fa -output masking/genome.repseq.fa -freq masking/genome.freq
# The file masking/genome.freq contains a list of ostensible repeat sequences.
cat masking/genome.repseq.fa | filter-stage-1.prl > masking/genome.repseq.f1.fa
# Remove repeats detected by RepeatScout that are too short or of low complexity.
cat masking/genome.repseq.f1.fa | filter-stage-2.prl --cat=masking/genome.fa.out > masking/genome.repseq.f2.fa
# filter out repeats which do not occur often enough.
The genome.repseq.f2.fa file will be used in RepeatMasker as repeat library.
The alignment tool STAR needs a hard-masked genome, whereas AUGUSTUS and BRAKER need it soft-masked, we generate both a soft-masked and a hard-masked genome.
RepeatMasker data/genome.fa -e ncbi -lib masking/genome.repseq.f2.fa -dir masking
mv masking/genome.fa.masked masking/genome.fa.hardmasked
RepeatMasker data/genome.fa -e ncbi -lib masking/genome.repseq.f2.fa -dir masking -xsmall
mv masking/genome.fa.masked masking/genome.fa.softmasked
BRAKER1 (GeneMark-ET and AUGUSTUS) can use genome and RNA-seq data to automatically generate full gene structure annotations in novel genome. BRAKER2 is the extension of BRAKER1 and can perform fully automated training of gene predition tools GeneMark-ET and AUGUSTUS from from RNA-Seq and/or protein homology information. BRAKER1 and BRAKER2 are executed by the same script braker.pl. For GeneMark-EX and AUGUSTUS, softmasking genome leads to better results.
There are several possible pipeline for BRAKER.
- Only genome file. This approach has lower prediction accuracy than all other described pipelines.

braker.pl --genome=genome.fa --esmode --softmasking --cores N
- Genome and RNA-Seq file from the same species. This approach is suitable for short read RNA-Seq libraries with a good coverage of the transcriptome.

braker.pl --species=yourSpecies --genome=genome.fasta \
--bam=file1.bam,file2.bam
braker.pl --species=yourSpecies --genome=genome.fasta \
--hints=hints1.gff,hints2.gff
BRAKER will have following important output files.
- augustus.hints.gtf: Genes predicted by AUGUSTUS with hints from given extrinsic evidence. This file will be missing if BRAKER was run with the option --esmode.
- GeneMark-E*/genemark.gtf: Genes predicted by GeneMark-ES/ET/EP/EP+ in GTF-format. This file will be missing if BRAKER was executed with proteins of close homology and the option --trainFromGth.
Using the following module and PATHs before running BRAKER.
# Load modules
module load bioinfo-tools
module load braker/2.1.1_Perl5.24.1
module load augustus/3.2.3_Perl5.24.1
module load bamtools/2.5.1
module load blast/2.9.0+
module load GenomeThreader/1.7.0
module load samtools/1.8
module load GeneMark/4.33-es_Perl5.24.1
# Your Command
export AUGUSTUS_CONFIG_PATH=/home/ziqi/Ziqi_Genome_Analysis/result/8_braker_annotation/config
# copy config folder into my working folder and chmod +w.
export AUGUSTUS_BIN_PATH=/sw/bioinfo/augustus/3.4.0/snowy/bin
export AUGUSTUS_SCRIPTS_PATH=/sw/bioinfo/augustus/3.4.0/snowy/scripts
export GENEMARK_PATH=/sw/bioinfo/GeneMark/4.33-es/snowy
BRAKER is a complex software and requires lots of settings before running sucessfully. The first step is to copy the config folder of AUGUSTUS into my work directory and change the permission into writtenable. Set the AUGUSTUS_CONFIG_PATH into my work directory. AUGUSTUS_BIN_PATH and AUGUSTUS_SCRIPTS_PATH also need to be exported. The simliar operation is performed on GENEMARK_PATH.
One common bug is: The hints file is empty. Maybe the genome and the RNA-seq file do not belong together. This bug could Solved by using this code to delete whitespaces in the headers of fasta file.
cut -d ' ' -f1 closed_gaps.fasta.masked > new_genome.fa
BREAKER workflow in this project includes 3 steps:
- Generate a set of training genes, based on the RNA-seq data we prepared, using GeneMark
- Train AUGUSTUS with these training genes
- Predict genes using the RNA-seq data as hints using AUGUSTUS
EggNOG-mapper is a tool for fast functional annotation of novel sequences. It uses precomputed orthologous groups (OGs) and phylogenies from the eggNOG database to transfer functional information from fine-grained orthologs only. EggNOG-mapper can be used in the annotation of novel genomes, transcriptomes or even metagenomic gene catalogs.
It is available online, but it has file size restriction. So in this project, local version will be used for analysis.
A basic usage example:
emapper.py -i FASTA_FILE_PROTEINS -o test
IGV means Integrative Genomics Viewer, it is used for visualize the genome annotation result. It can show the coverage rate and sequencing depth of the genome, structure variants are also avaliable.
Example for the main window of IGV:

STAR (Spliced Transcripts Alignment to a Reference) is an aligner designed to specifically address many of the challenges of RNA-seq data mapping using a strategy to account for spliced alignments. STAR is better than other aligners because it has higher accuracy and mapping speed, but it needs huge memory. STAR performs a two-step process: Seed searching and Clustering, stitching, scoring.
Here is an simple code example:
STAR --runMode genomeGenerate --genomeDir star --genomeFastaFiles masking/genome.fa.hardmasked
STAR --genomeDir star --readFilesIn data/rna-seq.fa --outFileNamePrefix star/ --outSAMtype BAM SortedByCoordinate
When using STAR on genome with large number of references, it is required to specifiy parameter --genomeChrBinNbits. It has a default value of 18, chrBin is the size of the bins for genome storage and each chromosome will occupy an integer number of bins. When it comes to large contigs, this parameter need to be calculate by min(18, log2[max(GenomeLength/NumberOfReferences,ReadLength)]) and keep the integer number. This is important, because the wrong number for --genomeChrBinNbits might cause core dump error.
Htseq can be used to count how many reads map to each feature with a given file including aligned sequencing reads and a list of genomic features.
For different data, feature has different meaning. In the case of RNA-Seq, the features are typically genes, where each gene is considered here as the union of all its exons. For comparative ChIP-Seq, the features might be binding region from a pre-determined list.
Htseq provide three different modes:
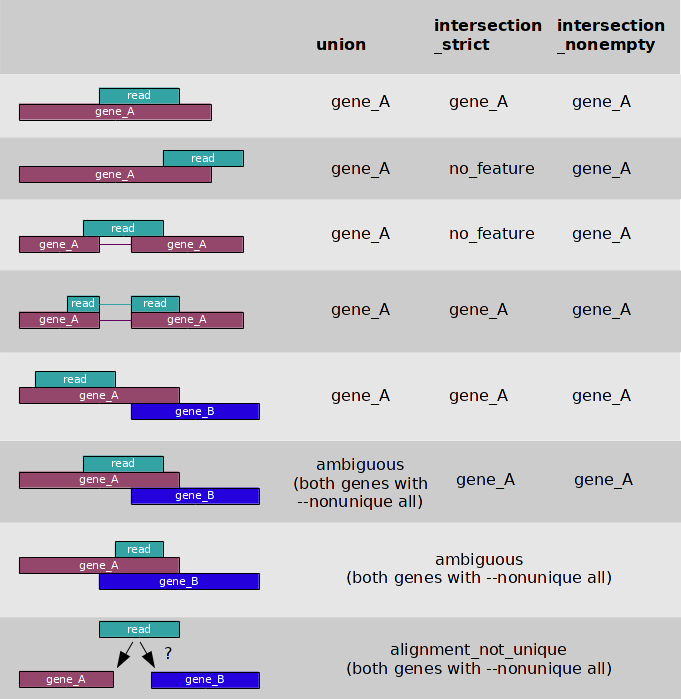
There is a simple code example. The script outputs a table with counts for each feature, followed by the special counters, which count reads that were not counted for any feature for various reasons.
htseq-count [options] <alignment_files> <gff_file>
# The <alignment_files> are one or more files containing the aligned reads in SAM format.
# The <gff_file> contains the features in the GFF format.
DEseq2 can be used to perform statistical analysis of count matrices for systematic changes between conditions and includes recommendations for producing count matrices from raw sequencing data.
DESeq2 package expects raw count data as obtained, e. g., from RNA-Seq or another highthroughput sequencing experiment, in the form of a matrix of integer values. DEseq2 requires to build DESeqDataSet matrix and the value in this matrix should be non-negative integers.
ddsFullCountTable <- DESeqDataSetFromMatrix(
countData = countdata,
colData = coldata,
design = ~ patient + treatment)
ddsFullCountTable
## class: DESeqDataSet
## dim: 63193 27
## exptData(0):
## assays(1): counts
## rownames(63193): ENSG00000000003 ENSG00000000005 ... LRG_98 LRG_99
## rowData metadata column names(0):
## colnames(27): SRR479052 SRR479053 ... SRR479077 SRR479078
## colData names(8): run experiment ... study sample
With the data object prepared, the DESeq2 analysis can be run with a single call to the function DESeq.
dds <- DESeq(dds)
Calling results without any arguments will extract the estimated log2 fold changes and p values for the last variable in the design formula.