3 conditions DEG - Kan-E/RNAseqChef GitHub Wiki
3 conditions DEG detects and visualizes differentially expressed genes by EBSeq multi-comparison analysis.
Note: Multiple comparison analysis takes 5-10 minutes.
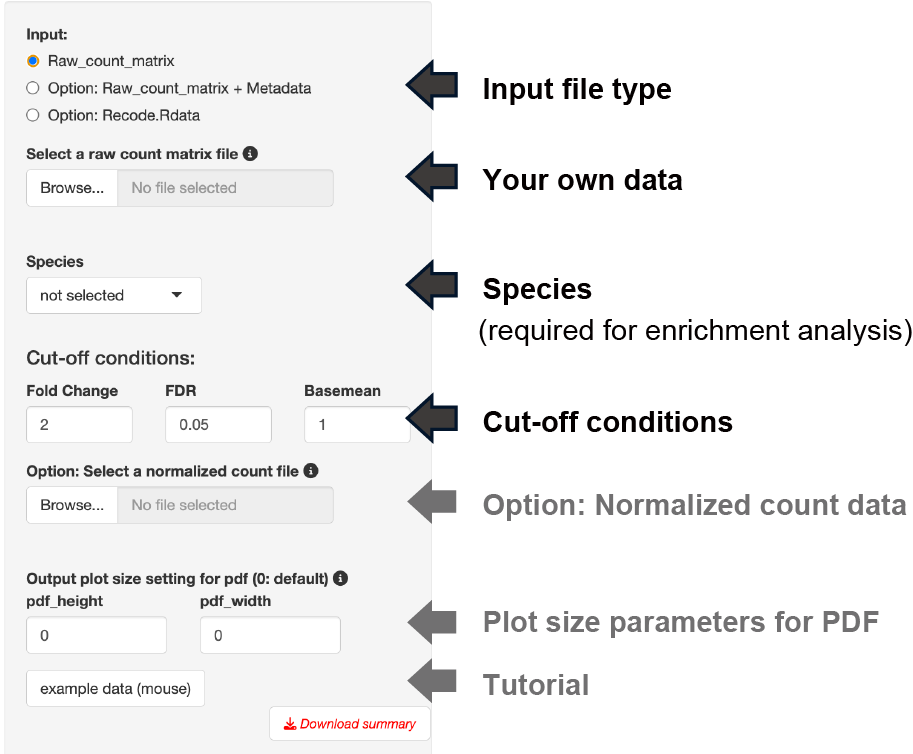
The input format and settings for the "3 conditions DEG" are the same as for pair-wise DEG.
If you have read the description of pair-wise DEG, you do not need to read the below.
Two types of raw count data formats can be used as input.
The analysis can only be performed with raw count data if the following conditions are fulfilled:
- A file consists of only three groups of data to be analyzed.
- The replication number is represented by the underline “_”.
- Do not use the underline "_" for anything else.
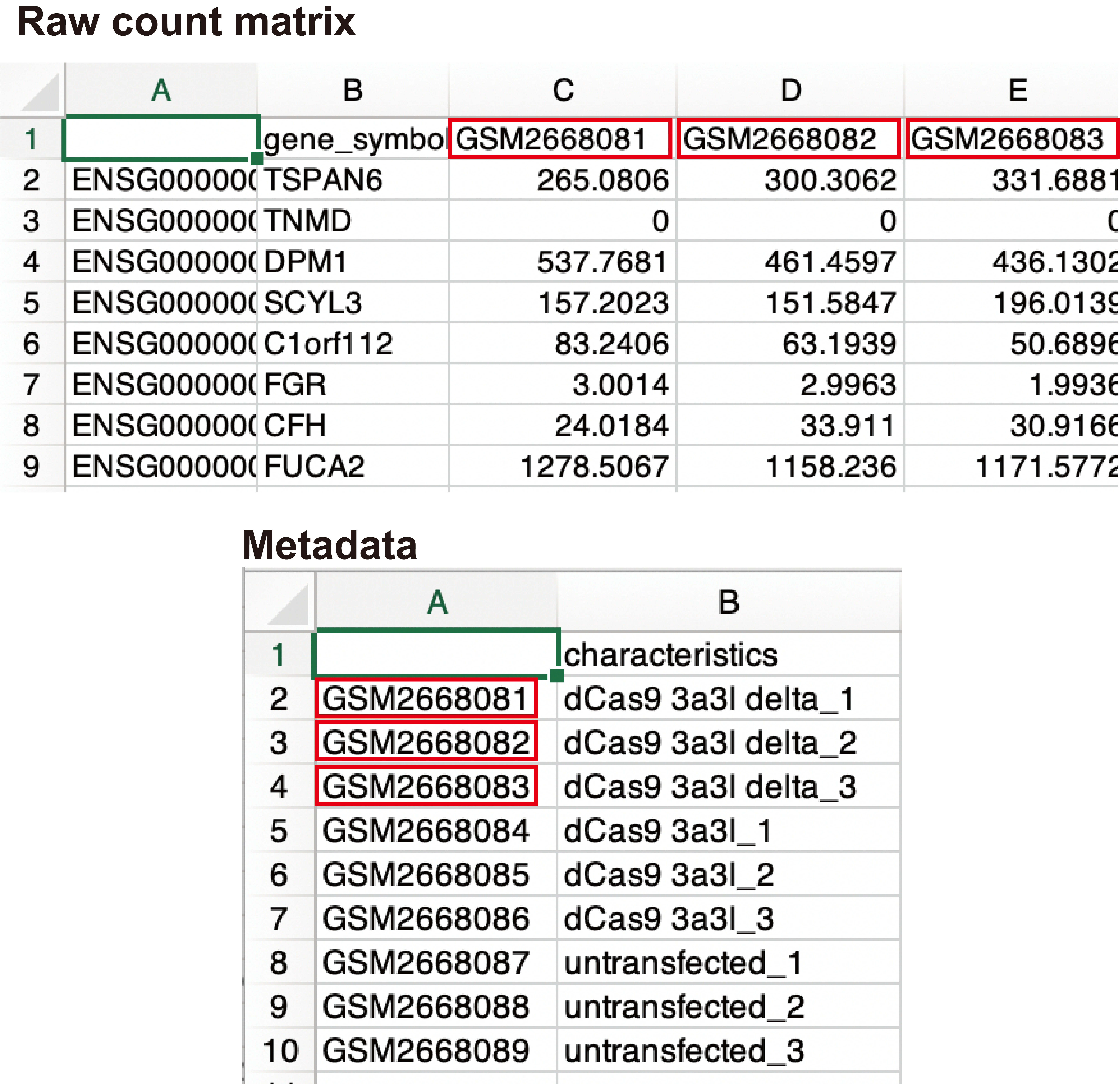
This format can be used if the above conditions are not fulfilled, for example, if the sample name is an accession number, or if the raw count data contain extra information that is not the subject of analysis.
Metadata must contain the following information:
- The first column is the sample names used in the raw count data.(e.g., accession number)
- The second column is the corresponding sample name that matches the sample name in the first column. (e.g. Control_1)
- The third and subsequent columns do not affect the analysis.
When you previously performed 3 conditions DEG and obtained "Recode.Rdata" file by clicking the 'Download summary' button, you can use this option to skip the time-consuming EBSeq analysis.
The following analysis is performed by selecting the dataset species.
- Conversion to gene symbols if the gene name is ENSEMBL ID
- Enrichment analysis
Three types of thresholds can be set: fold change, FDR, and base mean.
The base mean cut-off can be set using the uploaded normalized count data, such as TPM counts.
The y-axis of the boxplot can be displayed using the uploaded normalized count data.
Note: Uploading raw count data is not unnecessary.
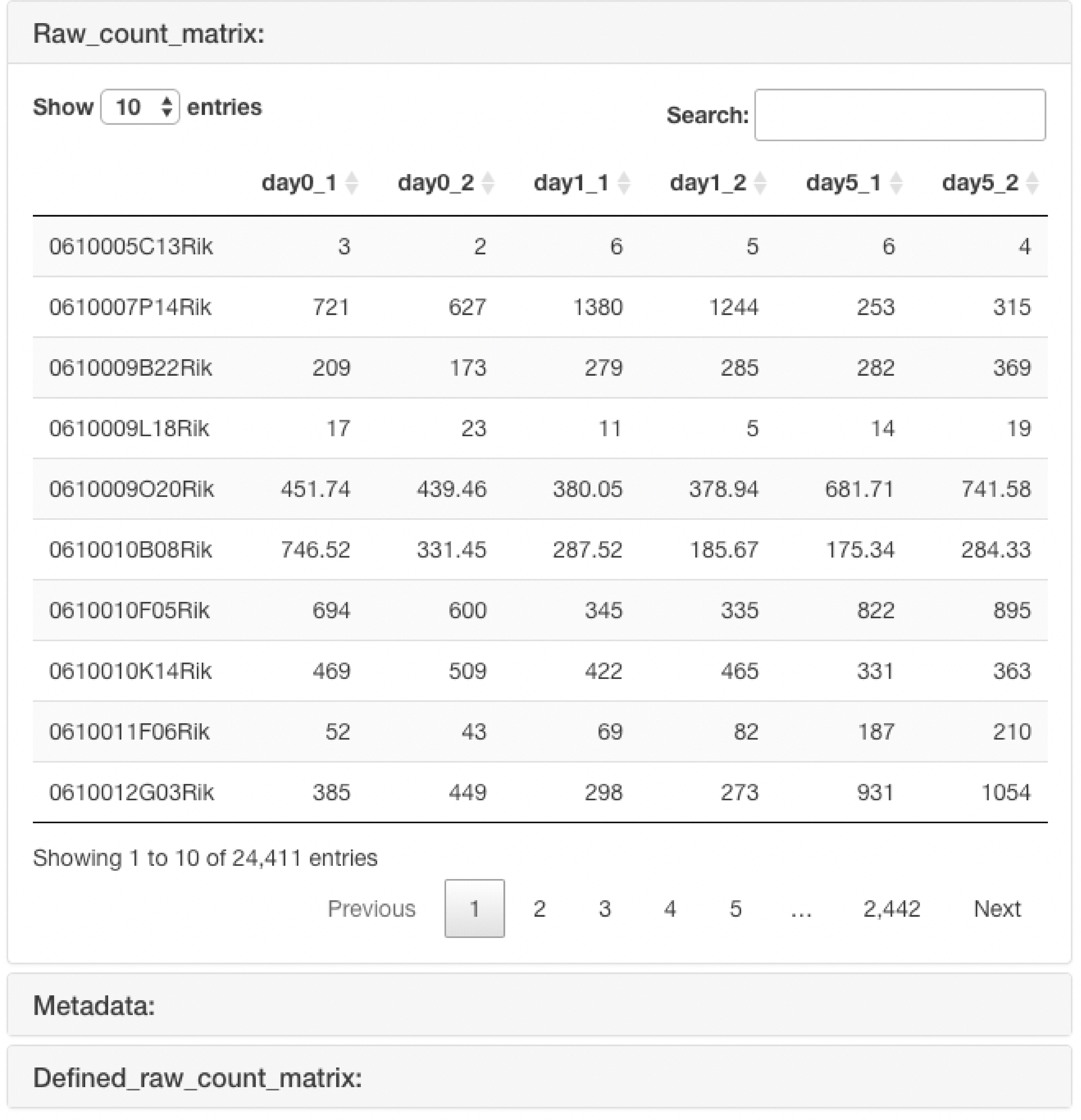
The uploaded raw count data are displayed.
In the case of the "Raw count data + metadata" format, the raw count data that is re-defined using the uploaded metadata.
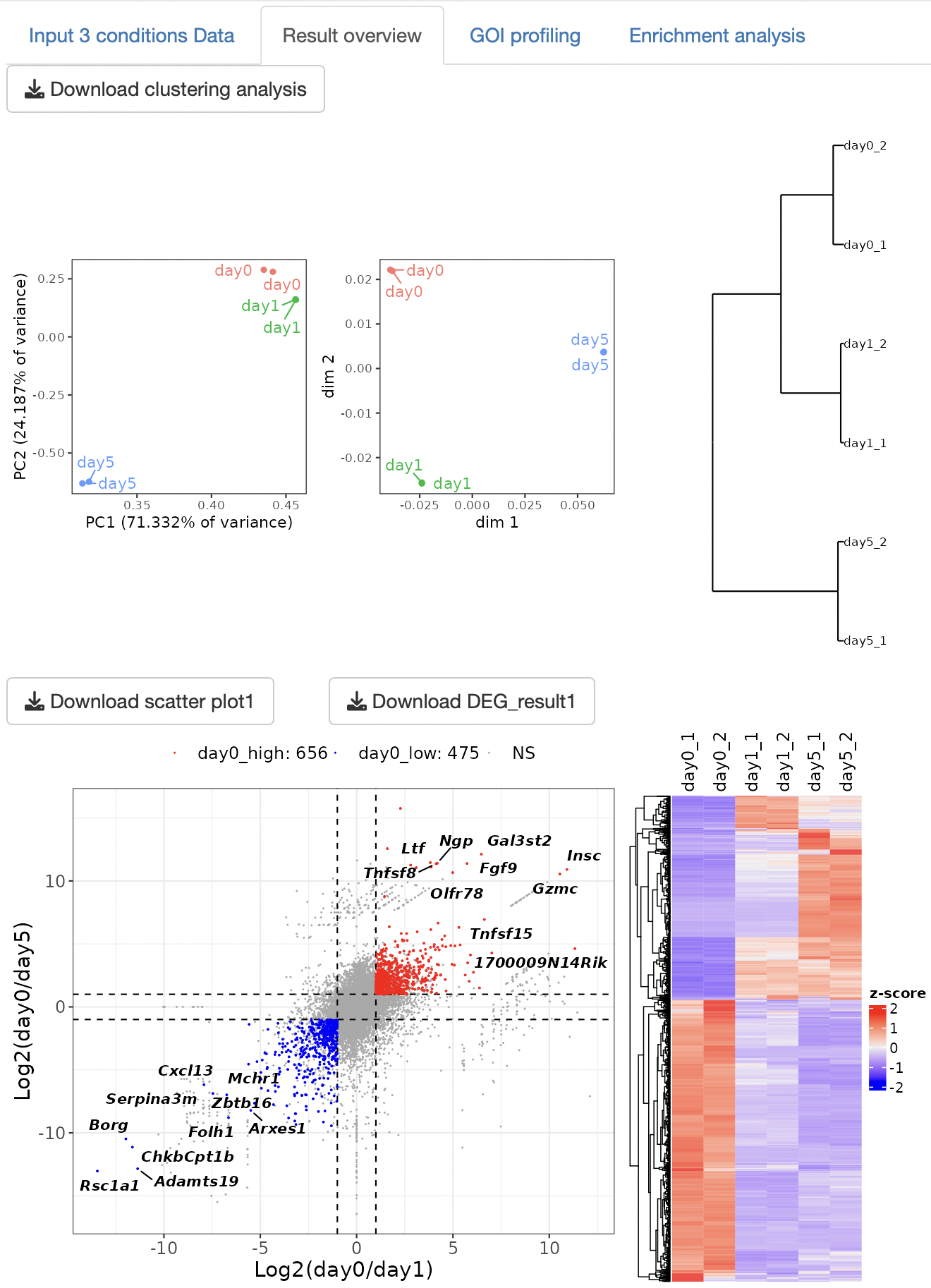
Three types of clustering analyses are performed: principal component analysis (PCA), multidimensional scaling (MDS), and hierarchical clustering with the ward.D2.
An EBSeq multi-comparison analysis is performed to detect the DEGs. Scatter plots and heatmaps are displayed as a result of the DEG analysis.
By visualizing DEGs under all three conditions, the characteristics of gene expression under each condition were extracted.
The result table data of the DEG analysis are displayed.
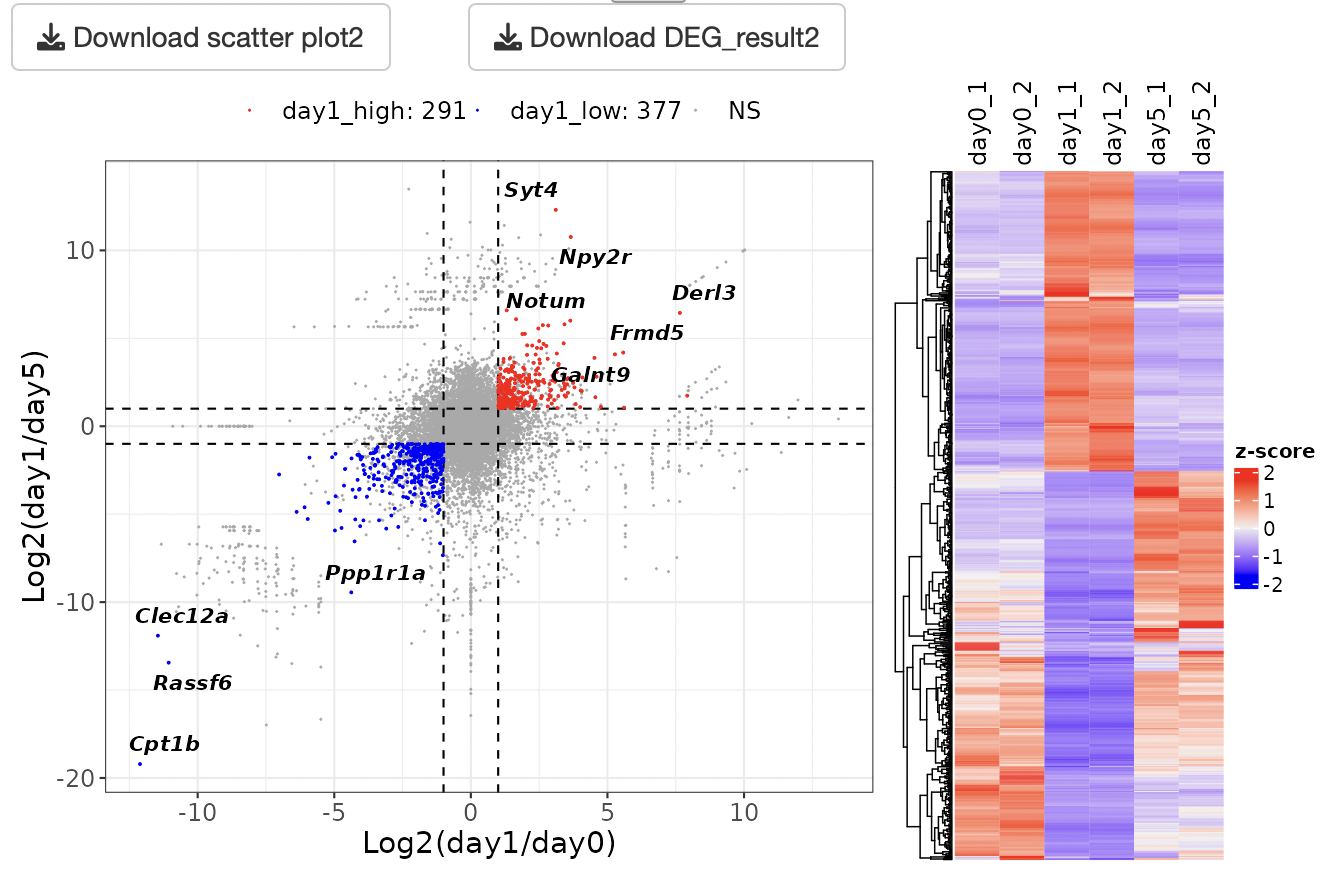
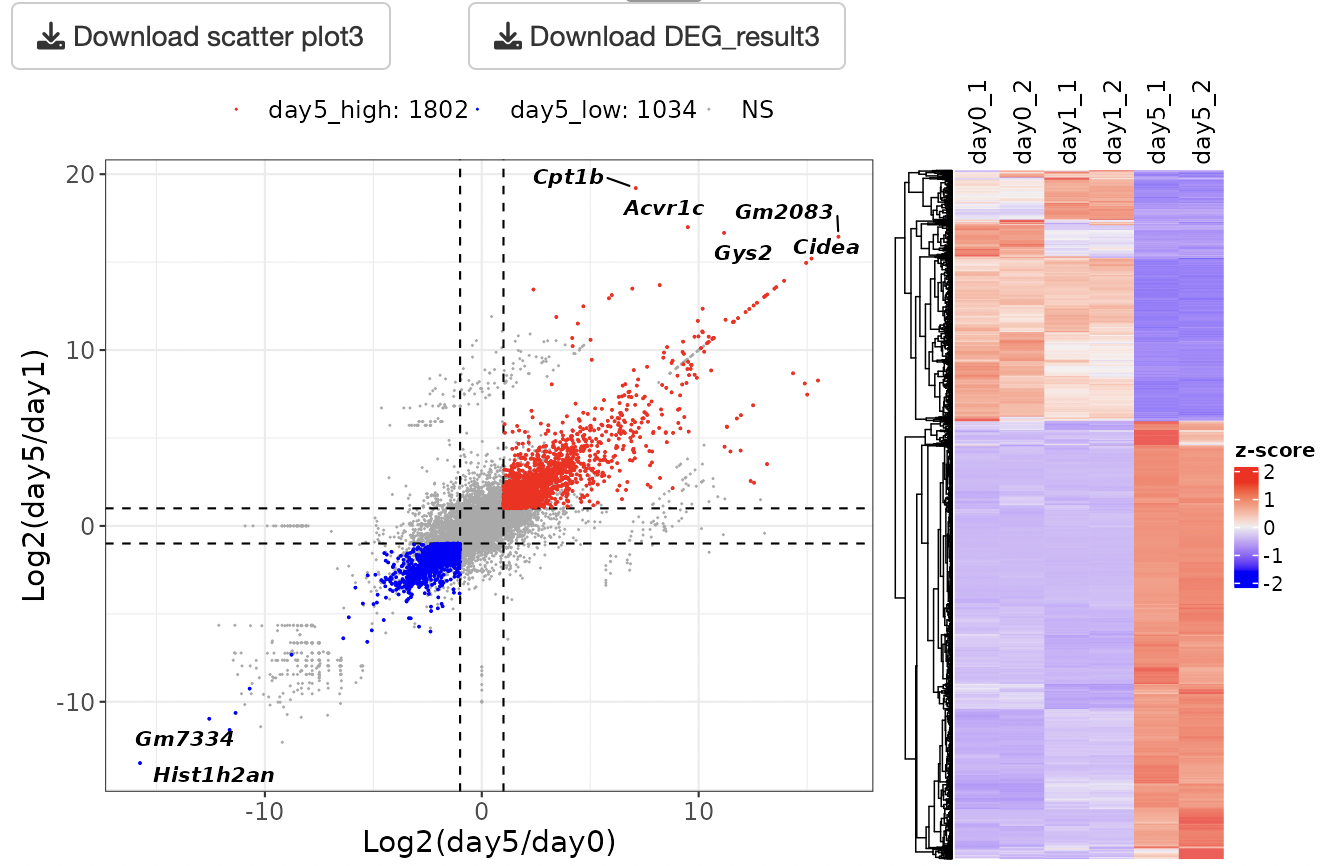
Scatter plot is shown.
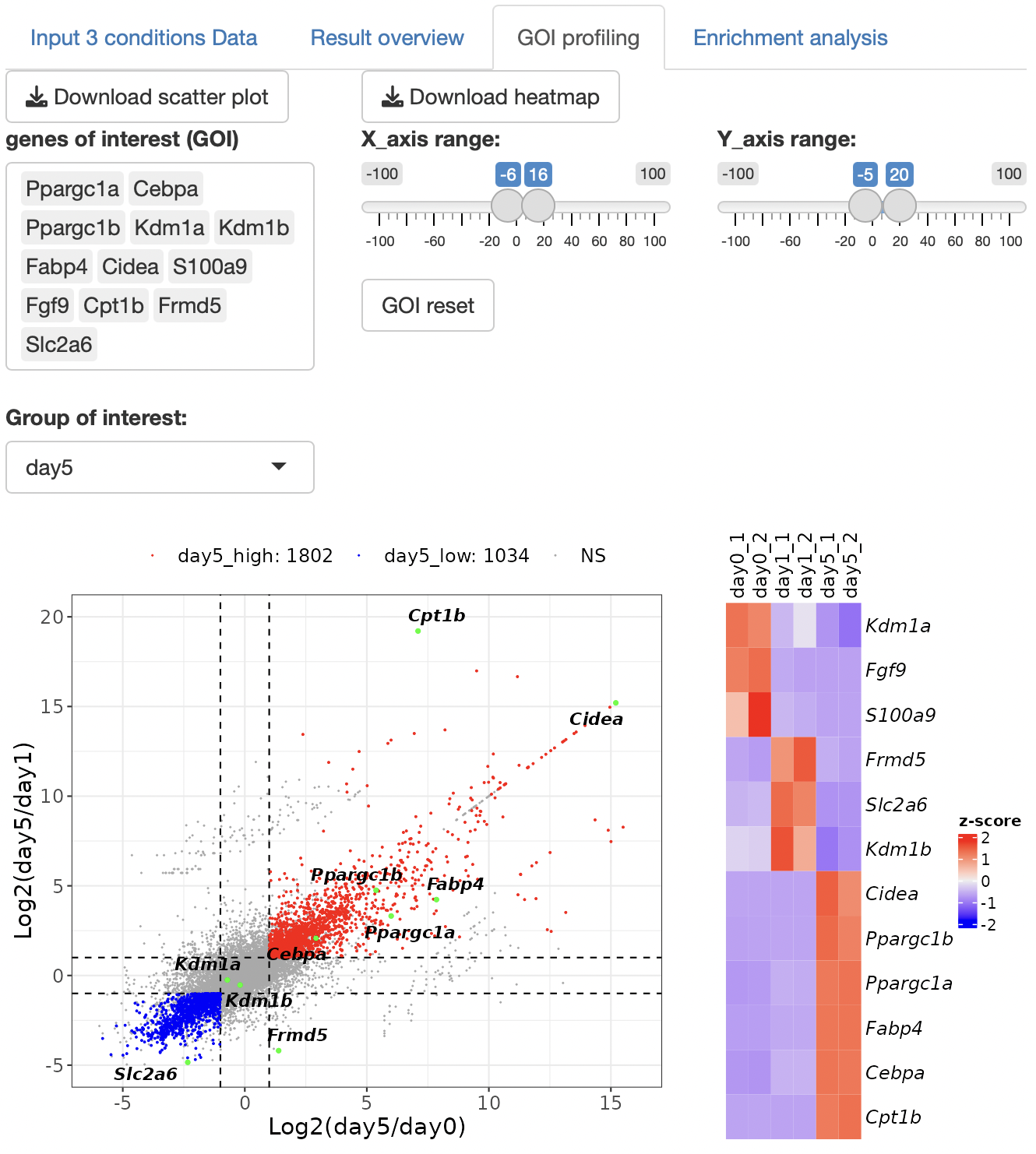
By selecting genes from the GOI list, only the GOI can be labeled among the points displayed in the volcano plot.
The x- and y-axis ranges could be freely changed by operating the slide bar.
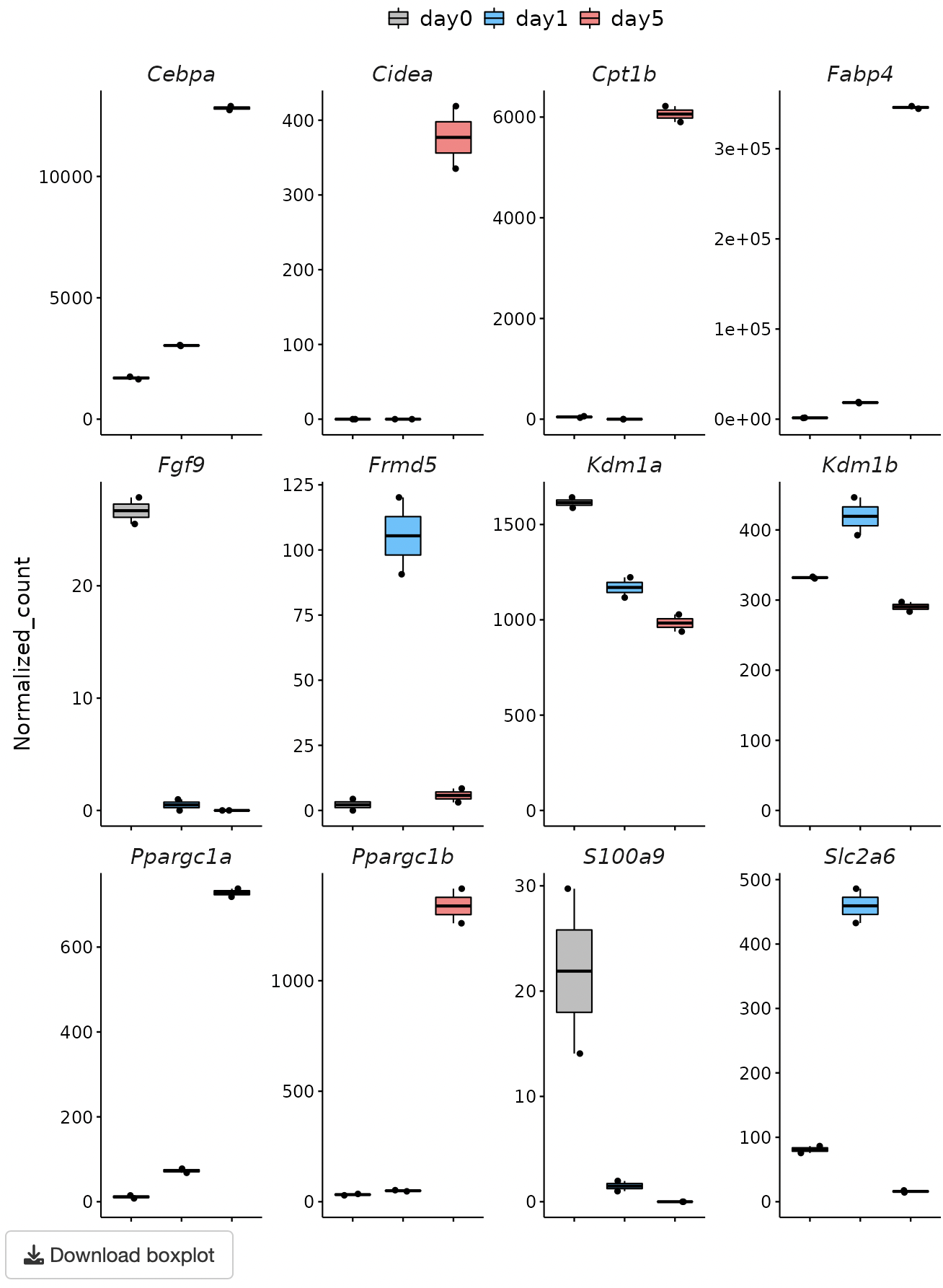
A heatmap and boxplot of the genes selected from the GOI list are displayed.
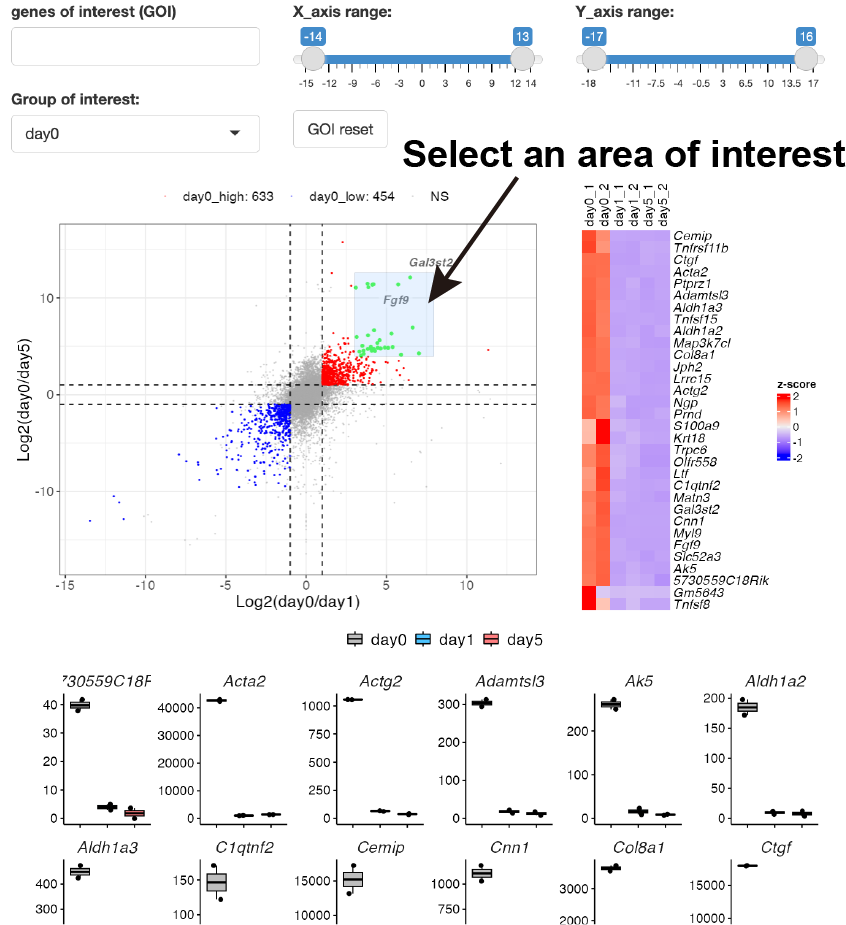
You can also select genes by drawing the box on the scatter plot.
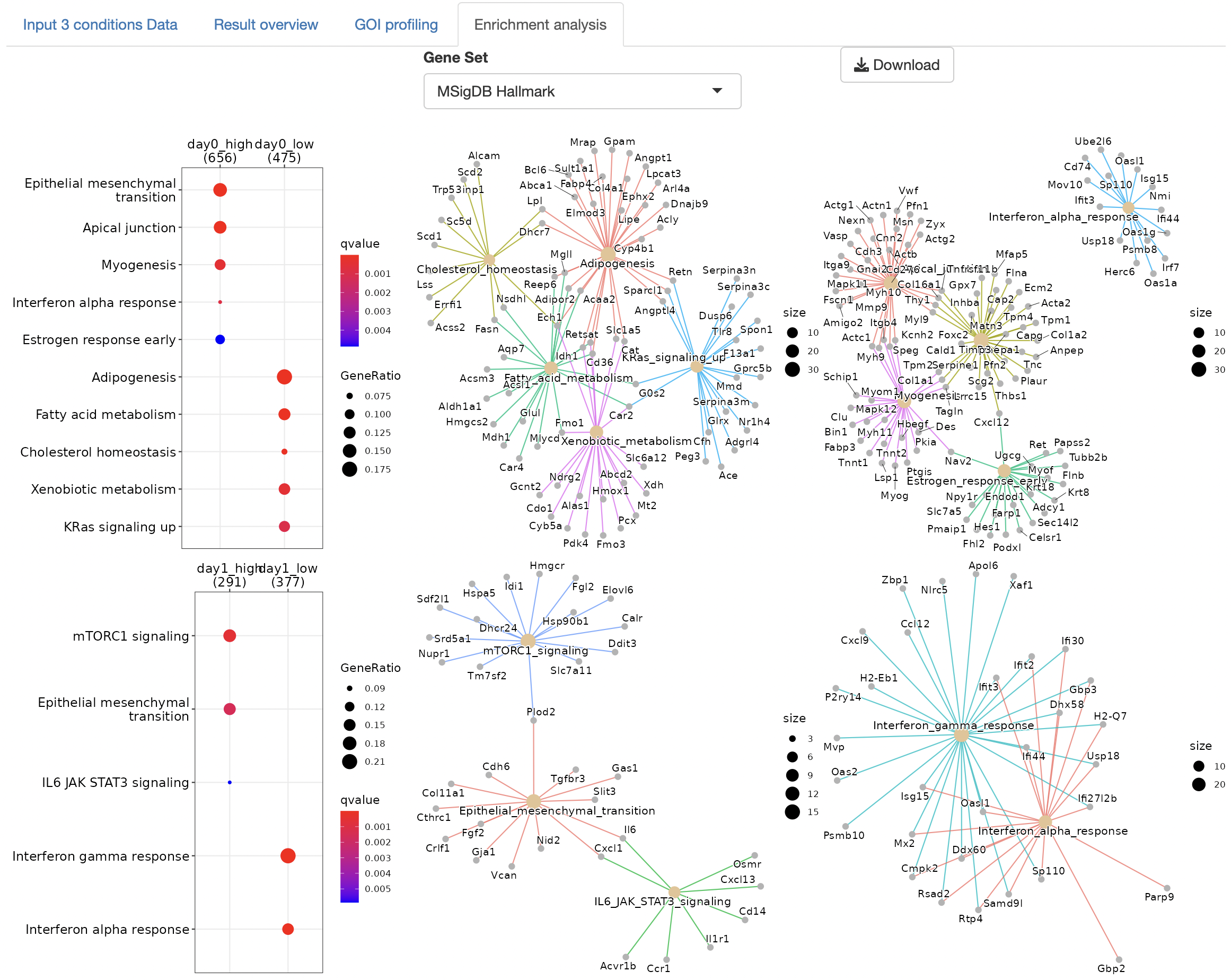
Overrepresentation analysis is performed to identify the function of DEGs.
The gene set can be selected from the following:
MSigDB hallmark gene
KEGG
Reactome
PID (Pathway Interaction Database)
BioCarta
WikiPathways
GO (biological process, cellular component, and molecular function)
Human phenotype ontology
DoRothEA regulon (activator)
DoRothEA regulon (repressor)
Transcription factor targets
miRNA target
Dotplots and gene-concept network, cnet, plots are displayed as the result.
Enrichment analysis depends on ClusterProfiler.