Contamination - KamilSJaron/k-mer-approaches-for-biodiversity-genomics GitHub Wiki
There are many ways how sequencing can go wrong, one very frequent pitfalls is contamination. This more often a problem for small species that are difficult to culture in sterile environments or even impossible to culture at all. Of course there are many forms of contamination - technical contamination (mixing samples in labs, spills etc), or biological contamination (content of a gut, other co-bionts of the hosts) or even somewhere in the middle with cases like environmental debris.
It is important to consider possible sources of contamination, because that might be helpful in considering the consequences for our sequencing run, but in general, it shows in k-mer spectra (at least with a rule of thumb).
The contamination was widely recognised in non-model genomics after the "scandal" regarding claims about extensive horizontal gene transfer in the tardigrade Hypsibius dujardini, but vast majority those were false positives due to contamination (Check Koutsovoulos et al. 2015 for details). Here, we will use the data from that paper to show how a dataset with so well characterised contamination looks like.
The k-mer spectra of H. dujardini was created using KMC from libraries ERR1147177, ERR1147178, and k=21. The k-mer specturm is available here. If you download the k-mer spectra, you can run genomescope on it
genomescope.R -i ERR1147177,ERR1147178.hist -o . -n Hduj -p 2
We will certainly obtain some model, but...
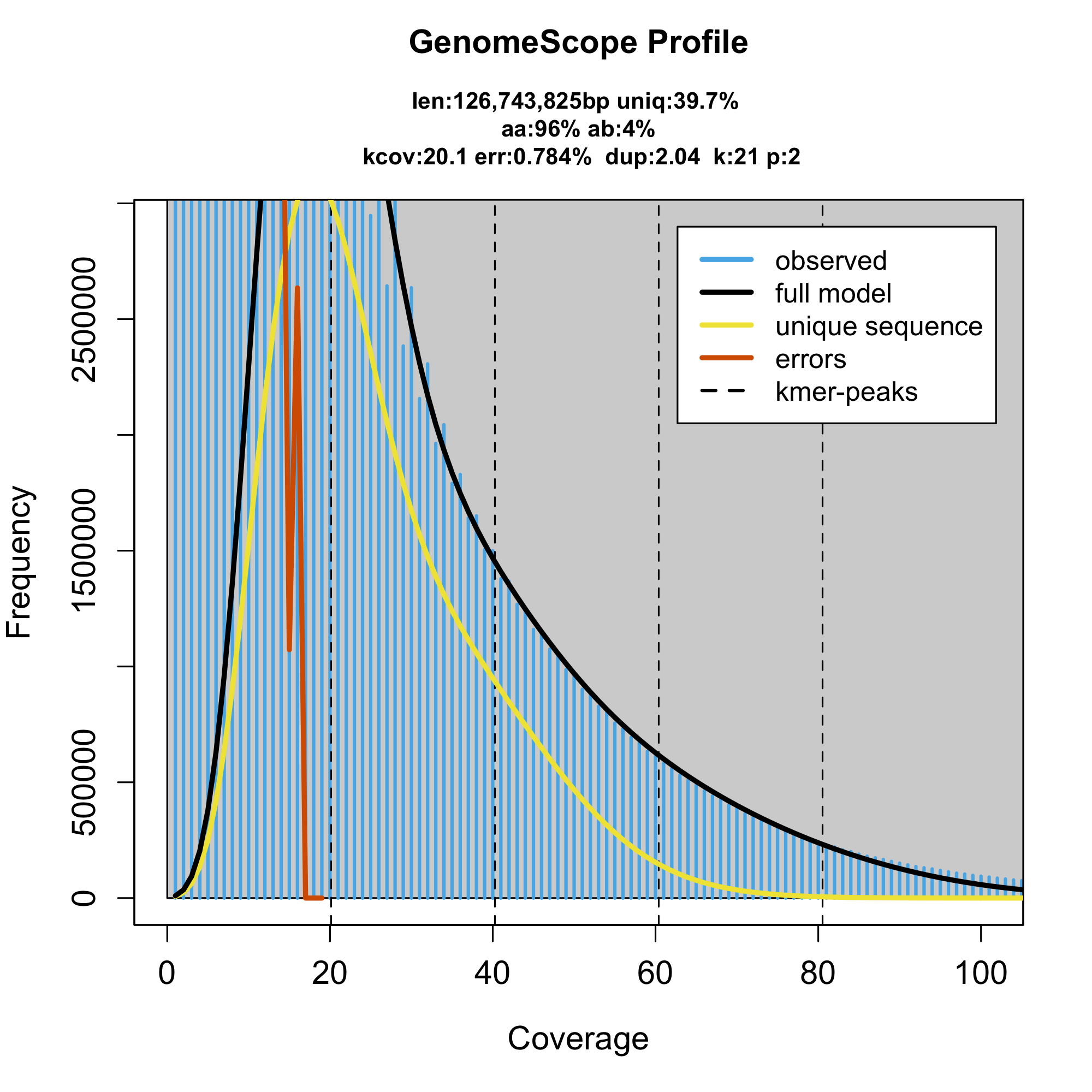
we also see that the k-mer spectrum does not show clear genomic peaks. This indicates that whatever we have sequenced, does not come from a single source and none of the sources is dominant enough, to show a separated genomic peak. Therefore, it is important to take the model with a giant grain of salt. We should largely ignore the estimated numbers that are rather meaningless. One that might indicate something is the estimated genome size, that is something that can help us guess where the issue is if we know approximately what to expect. Say, we know our genome is 400Mbp long, that means we simply don't have enough coverage because the true 1n coverage is a lot lot lower than what was estimated. However, if we thought our genome is 50Mbp, it indicates that it will be probably very well hidden among other things sequenced.
There are many ways how to deal with contamination and for a very thorough investigation, check Koutsovoulos et al. 2015. If we take the genome from that paper and use KAT (something like the line below should do the trick)
kat comp -o Hduj_kmer_profile_in_genome *.fastq.gz Hduj_genome.fa.gz
, we obtain following k-mer spectra
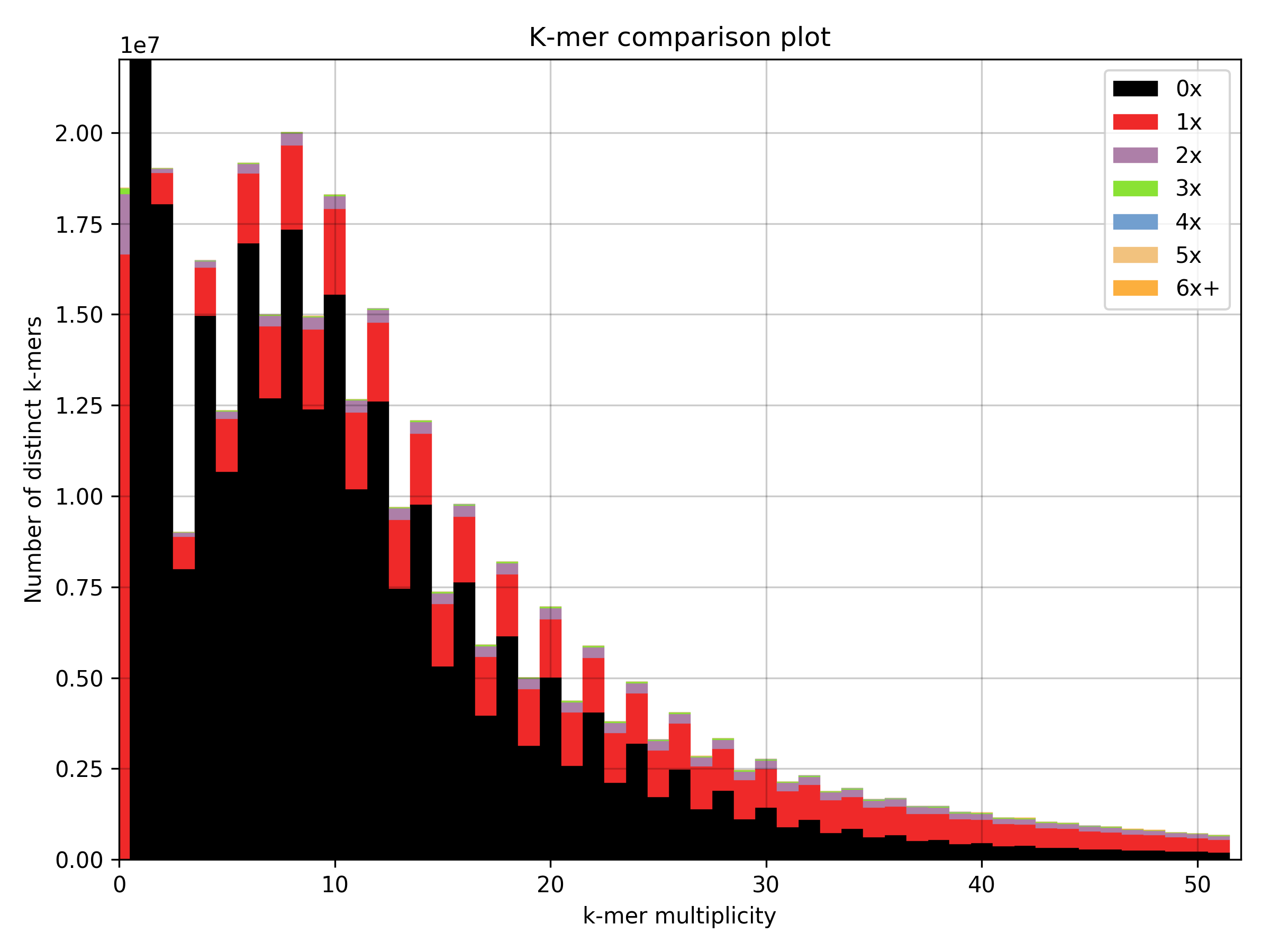
This shows that after the whole cleaning process of the assembly from all non-eukaryotic DNA, we ended up with only a small fraction of the original dataset. This is only to make the point that sometimes we indeed sequence a lot more other stuff than our actual target.
The lesson learned here - if I don't see peaks, I should expect a mess and not trust the fit model.