Running the script - Hoohm/CITE-seq-Count GitHub Wiki
How to use it
CITE-seq-count -R1 TAGS_R1.fastq.gz -R2 TAGS_R2.fastq.gz -t TAG_LIST.csv -cbf X1 -cbl X2 -umif Y1 -umil Y2 -hd 2 -o Result.tsv -wl Barcode_whitelist.txt
The script is going to count the number of UMIs linked to an antiobdy from your CITE-Seq experiment. PCR replicates are not counted multiple times.
If you are using the regex, you can check that it's working properly by using the -n option. This will run the script on only n reads.
You can always get a quick look at the options running the command CITE-seq-Count. This will print to std the list of arguments and their usage.
Here is an image quickly explaining the expected structure of read1 and read2 from the sequencer.
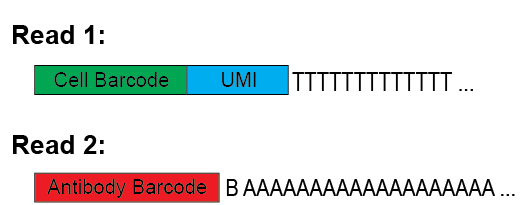
Options
You can find a description of each option bellow.
INPUT
[Required] Read1 fastq file location in fastq.gz format. Read 1 typically contains Cell barcode and UMI.
-R1 READ1_PATH.fastq.gz, --read1 READ1_PATH.fastq.gz
[Required] Read2 fastq file location in fastq.gz. Read 2 typically contains the Antibody barcode.
-R2 READ2_PATH.fastq.gz, --read2 READ2_PATH.fastq.gz
[Required] The path to the csv file containing the antibody barcodes as well as their respective names.
-t tags.csv, --tags tags.csv
Example of antibody file structure:
ATGCGA,First_tag_name
GTCATG,Second_tag_name
GCTAGTCGTACGA,Third_tag_name
GCTAGGTGTCGTA,Forth_tag_name
BARCODING
Positions of the cellular and UMI barcodes.
[Required] First nucleotide of cell barcode in read 1. For Drop-seq and 10x Genomics this is typically 1.
-cbf CB_FIRST, --cell_barcode_first_base CB_FIRST
[Required] Last nucleotide of the cell barcode in read 1. For 10x Genomics this is typically 16. For Drop-seq this depends on the bead configuration, typically 12.
-cbl CB_LAST, --cell_barcode_last_base CB_LAST
[Required] First nucleotide of the UMI in read 1. For 10x Genomics this is typically 17. For Drop-seq this is typically 13.
-umif UMI_FIRST, --umi_first_base UMI_FIRST
[Required] Last nucleotide of the UMI in read 1. For 10x Genomics this is typically 26. For Drop-seq this is typically 20.
-umil UMI_LAST, --umi_last_base UMI_LAST
Example: Barcodes from 1 to 16 and UMI from 17 to 26, then this is the input you need:
-cbf 1 -cbl 16 -umif 17 -umil 26
FILTERING
Filtering for structure of the antibody barcode as well as maximum hamming distance.
[Optional] Look for sequences of TAGS in R2 adding a [TGC] + polyA tail. Not suited for 5" sequencing. Is not activated by default.
-l or --legacy
[Required] Maximum hamming distance allowed. This allows to catch antibody barcode that might have HAMMING_THRESH error compared to the real read.
-hd HAMMING_THRESH, --hamming-distance HAMMING_THRESH
Example:
If we have this kind of antibody barcode:
ATGCCAG
The script will be looking for ATGCCAG in R2
A HAMMING_TRESH of 1 will allow barcodes such as ATGTCAG, having one mismatch to be counted as valid.
There is a sanity check when for the HAMMING_TRESH value chosen to be sure you are not allowing too many mismatches and confuse your antibody barcodes. Mismatches on cell or UMI barcodes are discarded.
[Optional] Structure of the sequenced antibody-oligo tag as aregular expression. This filters potential PCR artifacts (without a polyA tail) from true ADT reads (containing a polyA tail). For 15 nucleotide antibody barcodes and an R2 of at least 21 nucleotides this would be "^[ATGC]{15}[TGC][A]{6,}". Now optional, only for power users.
-tr TAG_REGEX, --TAG_regex TAG_REGEX
Example:
"^[ATGC]{15}[TGC][A]{6,}"
This will look at Read2 and look for this following structure:
^ Start of the read.
[ATGC]{15} Look for 15 base that have an A, T, C or G. This will be the antibody barcoding
[TGC] Look for one base of T, G or C. This base is important because it ensures we don't have the start of the polyA tail. Not all protocols use this base.
[A]{6,} Look for a sequence of 6 or more of base A. This is the polyA tail that we expect at the end of the antibody barcode. Only for 3prime protocols.
OUTPUT
You have to choose either the number of cells you expect or give it a list of cell barcodes to retrieve.
Number of expected cells from your run. This will reduce the size of the output matrix. It will catch 30% more than what you give to be sure to catch all cells.
-cells CELLS, --expected_cells CELLS
[OPTIONAL] If you have a whitelist of barcodes produced by the cDNA data, you are using a well-plate based protocol or any known whitelist, you can use this list to only extract TAGS matching those barcodes. Path to a txt file containning a whitelist of barcodes with no header.
-wl WHITELIST, --whitelist WHITELIST
Example:
ATGCTAGTGCTA
GCTAGTCAGGAT
CGACTGCTAACG
[Required] Path to the result matrix.
-o OUTFILE, --output OUTFILE
Cell barcodes as columns and antibody barcodes as rows.
OPTIONAL
Select first N reads to run on instead of all. This is usefull when trying to test out a regex pattern to be sure it's written properly before running the whole dataset.
-n FIRST_N, --first_n FIRST_N