Ntuple production & post processing - HEP-KBFI/tth-htt GitHub Wiki
The CRAB submission should be done in a clean environment. If you have more packages (e.g. ttH packages) in your CMSSW area, it would increase the job payload which will slow down the job (re)submission substantially. The jobs might never be submitted if the payload (which is the collection of all binary and data files in your CMSSW area) exceeds 100MB.
Dedicated CMSSW area in CRAB submission ensures that the jobs are submitted with the intended state of the CMSSW software.
# source exactly in this order
# on SL6 (deprecated)
#source /cvmfs/cms.cern.ch/crab3/crab_slc6.sh # deprecated
#source /cvmfs/cms.cern.ch/crab3/crab_slc7.sh # deprecated
source /cvmfs/cms.cern.ch/crab3/crab.sh prod
# on SL6 (deprecated)
#source /cvmfs/grid.cern.ch/emi-ui-3.17.1-1_sl6v2/etc/profile.d/setup-ui-example.sh
# on Centos7:
source /cvmfs/grid.cern.ch/umd-c7ui-latest/etc/profile.d/setup-c7-ui-example.sh
source /cvmfs/cms.cern.ch/cmsset_default.sh
# create a dedicated CMSSW area to submit the jobs
mkdir ~/submission_area
cd $_
cmsrel CMSSW_10_2_10
cd $_/src/
cmsenv
# pull our NanoAOD fork
git init
git config core.sparseCheckout true
echo -e 'PhysicsTools/NanoAOD/*\nDataFormats/MuonDetId/*\nRecoTauTag/RecoTau/*\n' > .git/info/sparse-checkout
git remote add origin https://github.com/HEP-KBFI/cmssw.git
git fetch origin
git checkout production_legacy_v5
git pull
# important
git clone -b production_legacy_v9 https://github.com/HEP-KBFI/tth-nanoAOD $CMSSW_BASE/src/tthAnalysis/NanoAOD
# compile
cd $CMSSW_BASE/src
scram b -j 8When creating the grid proxy, make sure that you keep it open for a long time so you don't have to rerun the same command multiple times to keep it open:
voms-proxy-init -voms cms -valid 192:00By default the proxy would be open 24 hours (I think), but the above command keeps it open for 192 hours. If the proxy dies, then all your jobs die (including the idling ones).
To test if you can write to Tallinn servers via grid proxy, run:
crab checkwrite --site=T2_EE_Estonia
You should see "success" or something to that effect on your screen.
Before you submit anything, create ~/crab_projects:
mkdir ~/crab_projectsThe job submission is done with launchall_nanoaod.sh script.
Even though the script is globally available, it assumes that it's executed in:
cd $CMSSW_BASE/src/tthAnalysis/NanoAODThe script is sort-of interactive as it might ask the following questions:
Sure you want to run NanoAOD production on samples defined in ...? [y/N]
Sure you want to generate config file: ...? [y/N]
Submitting jobs? [y/N]
These questions serve as the final frontier before the jobs are submitted.
If you don't spot any problems with the generation of the config files, just press y character and the script proceeds.
Here's what the script does:
-
guesses the list of datasets based on the command line arguments. No guess is made if the user has provided the list explicitly with
-foption; -
generates NanoAOD configuration files based on the command line arguments. Multiple MiniAOD production campaigns need to be distinguished with the
-e <era>option. Different MiniAOD types (data, FullSim MC, FastSim MC) determined with-j <type>option also imply different NanoAOD configuration. When running on data, golden JSON is automatically applied. There are also more options like:-
-tto set the number of processing threads (defaults to single thread); -
-pto publish the datasets (defaults no publication); -
-Tto skim events by OR of all HLT paths needed by the analysis (default no skimming); -
-rto modify logging frequency (defaults to once per 1000 events); -
-nto change the number of events produced by a CRAB job (defaults to 100'000 events); -
-vto give a specific name to the CRAB tasks (defaults toera_date).
All default values are suitable for NanoAOD production on the grid, so there's no need to change them.
-
-
generates configuration files in case there's a dataset that has at least one file block containing more than 100'000 luminosity blocks. These kind of datasets cannot be processed with the
EventAwareLumiBasedoption -- CRAB servers automatically refuse to submit them, onlyFileBasedsplitting option possible. However, some of these datasets may contain too large files and cannot be processed in a single CRAB job without exceeding the default time limit of 21h 55mins. To overcome that, multiple configuration files need to be generated to process one MiniAOD file in multiple jobs. For instance, if a CRAB job processes 100'000 but the largest file in the dataset has 250'000 events, three configuration files are generated, where the first one processes the first 100'000 events, the second one 100'000th to 200'000th event and the third one 200'000th to 250'000th event; -
checks if the grid proxy is open long enough;
-
submits the job, one dataset at a time.
Notice that in some cases -s option may be used.
This option is there to give the generated NanoAOD configuration file a different name, so that they wouldn't be overwritten by the next command.
The submission alone may take a few hours. Unfortunately, it's not possible to redirect the screen output to a file because CRAB server might ask you the password to your grid certificate interactively (even though you had entered the password recently when opening the grid proxy).
The CRAB log files will be stored in the subdirectories of ~/crab_projects.
# to submit data
launchall_nanoaod.sh -f test/datasets/txt/datasets_data_2016_17Jul18.txt -j data -e 2016v3
# to submit MC
launchall_nanoaod.sh -j mc -e 2016v3# to submit data
launchall_nanoaod.sh -f test/datasets/txt/datasets_data_2017_31Mar18.txt -j data -e 2017v2
# to submit MC
launchall_nanoaod.sh -j mc -e 2017v2# to submit data, runs A, B, C
launchall_nanoaod.sh -f test/datasets/txt/datasets_data_2018_17Sep18.txt -j data -e 2018
# to submit data, run D
launchall_nanoaod.sh -f test/datasets/txt/datasets_data_2018_17Sep18.txt -j data -e 2018prompt
# to submit MC:
launchall_nanoaod.sh -j mc -e 2018We want to process fewer number of events in NanoAOD Ntuples because later analyzing them will become a problem if the Ntuples contain too many events.
# to submit HH multilepton samples (no 2018)
launchall_nanoaod.sh -f test/datasets/txt/datasets_hh_multilepton_mc_2016_RunIISummer16MiniAODv3.txt -j mc -e 2016v3 -n 50000 -s hh_multilepton
launchall_nanoaod.sh -f test/datasets/txt/datasets_hh_multilepton_mc_2017_RunIIFall17MiniAODv2.txt -j mc -e 2017v2 -n 50000 -s hh_multilepton
# to submit HH bbww samples
launchall_nanoaod.sh -f test/datasets/txt/datasets_hh_bbww_mc_2016_RunIISummer16MiniAODv3.txt -j mc -e 2016v3 -n 50000 -s hh_bbww
launchall_nanoaod.sh -f test/datasets/txt/datasets_hh_bbww_mc_2017_RunIIFall17MiniAODv2.txt -j mc -e 2017v2 -n 50000 -s hh_bbww
launchall_nanoaod.sh -f test/datasets/txt/datasets_hh_bbww_mc_2018_RunIIAutumn18MiniAOD.txt -j mc -e 2018 -n 50000 -s hh_bbwwlaunchall_nanoaod.sh -f test/datasets/txt/datasets_ttbar_mc_2016_RunIISummer16MiniAODv3.txt -j mc -e 2016v3 -s ttbar
launchall_nanoaod.sh -f test/datasets/txt/datasets_ttbar_mc_2017_RunIIFall17MiniAODv2.txt -j mc -e 2017v2 -s ttbar
launchall_nanoaod.sh -f test/datasets/txt/datasets_ttbar_mc_2018_RunIIAutumn18MiniAOD.txt -j mc -e 2018 -s ttbarIn the examples below it is assumed that 2017v2 jobs are submitted on May 31st.
First, you have to make sure that all jobs have been submitted and that there are no errors.
The easiest way is to loop over the subdirectories in your ~/crab_projects area and run crab status on them like so:
for d in ~/crab_projects/crab_2017v2_2019May31_*; do crab status -d $d; doneYou may want to dump it to a file so because running crab status hundreds of times may take a while:
for d in ~/crab_projects/crab_2017v2_2019May31_*; do crab status -d $d; done &> crab_status.log
Initially, you want to wait for some jobs to fail before you can resubmit them. Once the Ntuple production gets going, you can resubmit them automatically like so:
while true; do for d in ~/crab_projects/crab_2017v2_2019May31_*; do crab_resubmit.sh -d $d; done; echo "Sleeping for 86400 seconds"; echo `date`; sleep 86400; done
The one-liner automatically resubmits all failed jobs every 24 hours.
There are currently two web site available for monitoring:
- http://dashb-cms-job.cern.ch/dashboard/templates/task-analysis
- https://monit-grafana.cern.ch/d/cmsTMGlobal/cms-tasks-monitoring-globalview
The first one will be deprecated very soon. Also, it doesn't display all CRAB tasks for some reason.
The second website is what we're supposed to migrate over to. Unfortunately, the website doesn't provide a pie chart summarizing the progress of all CRAB tasks. For this purpose we have a script that generates the pie chart by parsing the CRAB log files locally:
plot_crab_status.py -i ~/crab_projects -p crab_2017v2_2019May31_.* -o pie.pngExample (production of 193 data & MC datasets, roughly 4.8 billion events):
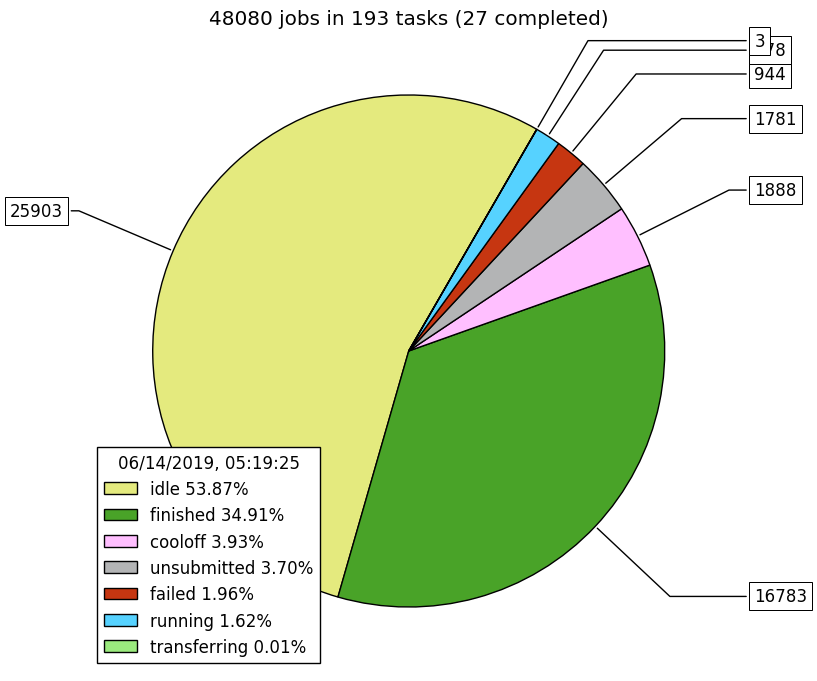
At the time of writing this text, there should be no obvious problems with the NanoAOD software, except for this issue where the 900th event in the MiniAOD file triggers a 500MB jump in memory consumption, probably due to "poor clustering" of this event. There are no workaround to this problem, unless maybe increasing the memory limit of the CRAB jobs.
Unfortunately, it's not possible to distinguish genuine memory overconsumption from "fake" overconsumption: the jobs may fail due to poor network conditions (or due to poor implementation of gsiftp/xrd protocol) that may cause a temporary spike in memory consumption. This in turn causes the job to exit with error code that corresponds to memory overconsumption instead of code that stands for file reading errors. The same argument applies to jobs that fail due to segmentation violation. The argument is supported by the fact that the errors are not reproducable interactively, the error logs oftentimes show warnings about network connection and that the jobs that have been resubmitted repeatedly have eventually succeeded.
The main reason why the jobs are idling and why many of them fail is because the datasets are replicated on a very few number of tier-2 (or lower) sites where CRAB jobs can run. The only "solution" is to wait for the datasets to be replicated on more sites, which will increase the probability for the jobs to start and finish successfully.
Raising the memory limit on CRAB jobs should be used as a last resort, eg. when most of the jobs have run at least once and failed due to genuinely hitting the memory cap. When the memory limit is raised, it significantly decreases the probability (or outright set it to zero) for the jobs to even start because there are very limited number of sites that can allocate more than 2000MB per job. Combined with limited availability of the datasets and the fact that it's not possible to turn off the locality option via CRAB commands, it follows that the number of cores in the job must be increased as well.
The full analysis software is needed to run post-production due to complicated dependencies. Here are the instruction for setting up the rest of the software.
NB! Different CMSSW area should be used than what was used to submit CRAB jobs, especially if the CRAB jobs are still running.
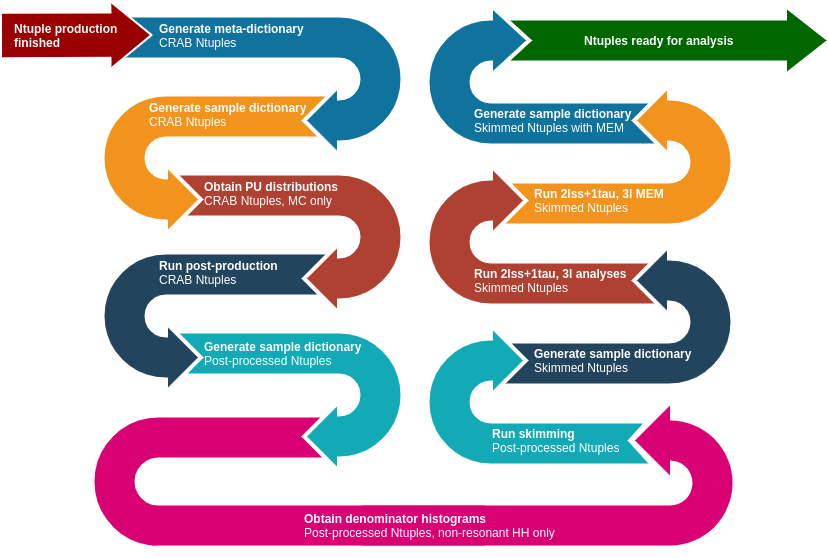
The post-production/post-processing can be split into three main steps which prepare for and execute:
- the actual post-processing:
- add new information to the Ntuples;
- filter out some objects that pass the loosest criteria needed in the analysis;
- skimming -- select events that contain enough number of objects;
- MEM computation -- relevant only in a few analyses.
Each of these steps consists of subtasks as shown in the diagram below. More details are given in the following sections.
The following files have to be produced before any post-processing can start in a given era and analysis:
- for MC:
- a text file that lists all MC DBS names;
- another text file containing list of sample names that cover the same phase space;
- a third text file for keeping the list of paths to the Ntuples produced with CRAB jobs;
- meta dictionary;
- sample dictionary;
- histograms for the true number of PU vertices in every sample individually;
- for data:
- meta dictionary;
- sample dictionary.
Preparing post-processing jobs for data is easier than that for MC, because only meta- and sample dictionaries are needed for this step.
First, the meta-dictionary can be simply generated with find_samples.py script which does the following:
- find all DBS names for data samples, given
- list of PD (eg EGamma, SingleMuon), enabled with
-poption, for a given era (-roption); - minimum CMSSW version the samples were produced with, specified with
-vkey; - a list of strings that the DBS names have to include (
+as prefix) or must not include (xas prefix) with-Coption; - if there are multiple (not invalid) candidates for a given PD and acquisition era, then the one that has more events is selected. All considered DBS names are shown on screen if debug output is enabled with
-Vflag;
- list of PD (eg EGamma, SingleMuon), enabled with
- find golden run ranges;
- only if Golden JSON is provided with
-goption; - the result is purely needed only for documentation purposes.
- only if Golden JSON is provided with
- compute integrated luminosity for each PD and acquisition era;
- implies that Normtags has been checked out, preferably in
$HOME(but can be overwritten with-Nkey); - is actually enabled with
-lflag; - as with golden run ranges, the result is needed only for documentation purposes.
- implies that Normtags has been checked out, preferably in
There are two delivarables:
- a plain text file (location determined with
-doption):- each line corresponds to one DBS name, followed by run range, golden run range, integrated luminosity, and integrated luminosity in the golden run range. The figures that correspond to the golden run range should be used in the documentation;
- lines starting with a hastag are comments, which include the actual command that generated the file itself;
- the text file is actually the one used as an input to
launchall_nanoaod.shscript that submits Ntuple production jobs to CRAB;
- meta-dictionary (location set by
-moption).
The meta-dictionary is a python dictionary that holds useful information about every dataset queried via dasgoclient such as:
- number of valid events (accurate, and a rounded up version for humans to read);
- number of MINIAOD files;
- production status of the sample (eg VALID or PRODUCTION);
- total size of the MINIAOD files;
- CMSSW version that was used in creating the MINIAOD files;
- last time the sample was modified in DBS.
Additional information, such as golden run ranges, sample names and sample categories are also stored in the dictionary. Sample name should not be confused with DBS name: the former is freely given but must be unique to each sample, while the latter is a fixed name. Sample category is basically a label that collects multiple samples under one label (eg TTZ refers to all samples of that process).
Finally, each entry in the meta-dictionary has a field named crab_string, which is a unique string present in the Ntuple paths.
The string is empty if no paths to the CRAB Ntuples are found, or if the list of paths was not supplied to find_samples.py with -c option.
This list can be generated from the CRAB directories, for instance
find_crab_directories.py \
-i ~/crab_projects ~anotherUser/crab_projects \
-o path/to/sample/locations.txt \
-c crab_2017v2 \
-t 30will save all paths to CRAB tasks that contain crab_2017v2 in their name and are at least 30% complete.
The results are found by parsing the log files in the CRAB directories (given by -i) and saved to the path specified with -o option.
As with the plaintext files, the meta dictionary also includes the command that generated the file itself. The meta-dictionary has to be generated for:
- both data and MC separately;
- every era;
- each analysis (eg ttH, HH multilepton and HH bbWW samples need separate meta dictionaries).
The point of having a "meta-dictionary" in the analysis is to maintain invariant set of information (as basic as sample categories) that is carried over to the next steps. Without this intermediate step, building the sample dictionaries would be much more complex than it already is.
Below are some examples that generated the meta-dictionaries (and accompanying text files) for each era in ttH analysis, and a figure showing excerpts from the generated output files:
# example A:
find_samples.py -V -v 9_4_9 -r 2016 -l -m python/samples/metaDict_2016_data.py \
-d ../NanoAOD/test/datasets/txt/datasets_data_2016_17Jul18.txt \
-p SingleElectron SingleMuon DoubleEG DoubleMuon MuonEG Tau \
-g ../NanoAOD/data/Cert_271036-284044_13TeV_23Sep2016ReReco_Collisions16_JSON.txt \
-C +17Jul2018 xDoubleEG/Run2016B-17Jul2018_ver1-v1 \
-c python/samples/sampleLocations_2016_nanoAOD.txt
# example B
find_samples.py -V -v 9_4_5_cand1 -r 2017 -l -m python/samples/metaDict_2017_data.py \
-d ../NanoAOD/test/datasets/txt/datasets_data_2017_31Mar18.txt -C +31Mar2018 \
-p SingleElectron SingleMuon DoubleEG DoubleMuon MuonEG Tau \
-g ../NanoAOD/data/Cert_294927-306462_13TeV_EOY2017ReReco_Collisions17_JSON_v1.txt \
-c python/samples/sampleLocations_2017_nanoAOD.txt
# example C
find_samples.py -V -v 10_2_4_patch1 -r 2018 -l -m python/samples/metaDict_2018_data.py \
-d ../NanoAOD/test/datasets/txt/datasets_data_2018_17Sep18_22Jan19.txt \
-p SingleMuon EGamma DoubleMuon MuonEG Tau \
-g ../NanoAOD/data/Cert_314472-325175_13TeV_17SeptEarlyReReco2018ABC_PromptEraD_Collisions18_JSON.txt \
-C +17Sep2018 \
+Run2018D-PromptReco +22Jan2019 \
x/SingleMuon/Run2018D-PromptReco-v2/MINIAOD \
x/EGamma/Run2018D-PromptReco-v2/MINIAOD \
-c python/samples/sampleLocations_2018_nanoAOD.txt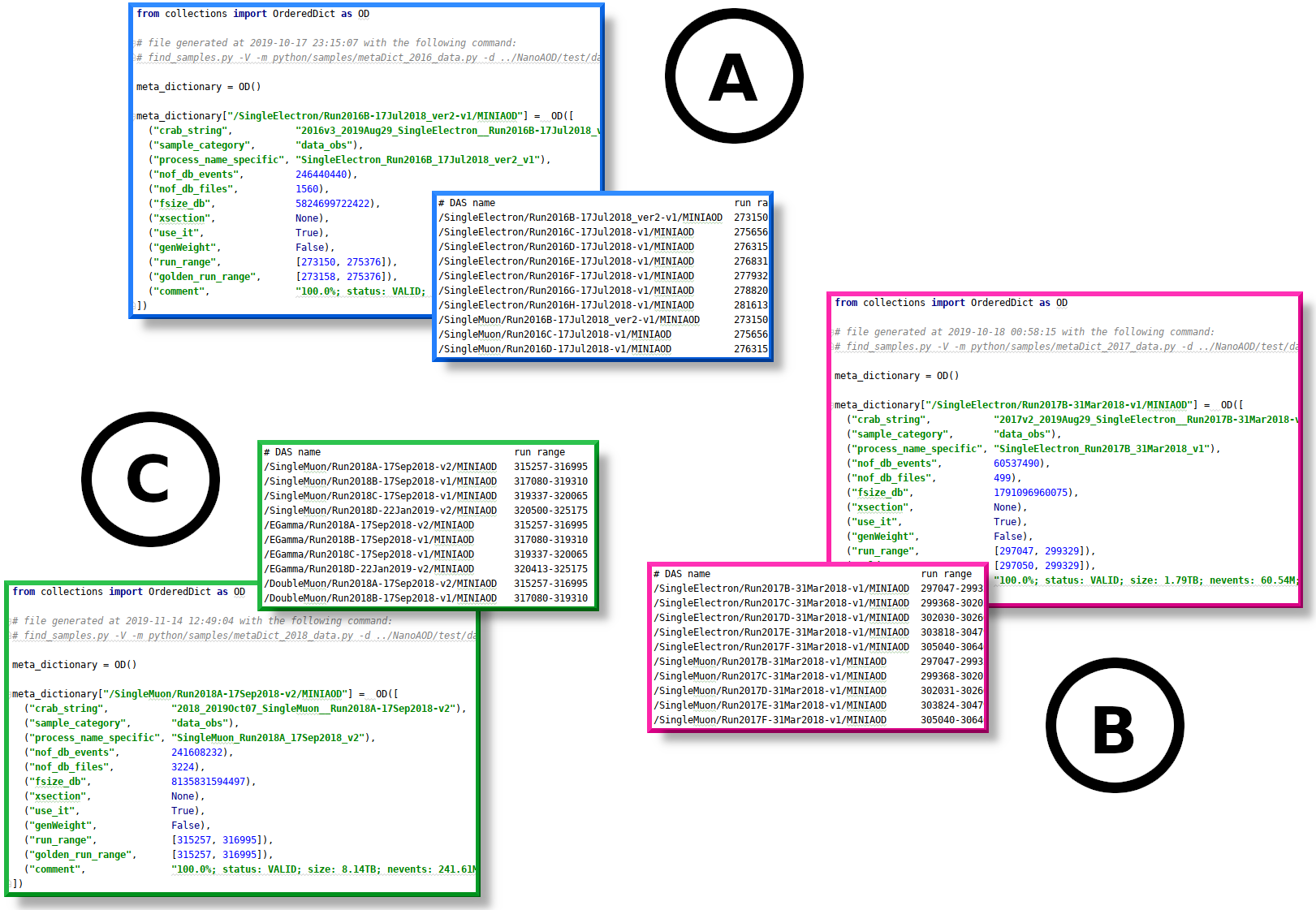
Generating a meta-dictionary for MC is much more complicated, because it's not sensible to "harvest" the needed datasets via dasgoclient.
Instead, the list of datasets has to be determined beforehand.
Furthermore, the samples that represent the same physics process/phase space must share the same name, category and cross section, regardless of which era these samples belong to.
This motivated the creation of JSON files that have the following nested structure:
- sample category (such as: ttH, TTZ, QCD);
- each sample category may contain multiple "sample sets" in order to differentiate them by MC generation. For instance
ttHJetToNonbb_M125_amcatnlosamples are NLO ttH samples andttHToNonbb_M125_powhegare LO ones. This separation is necessary, because the NLO and LO samples must not be mixed together at the analysis level; - each of such sample set is accompanied with:
- cross section value in 1/pb, order and origin / method of calculation;
- auxiliary information, such as the use case for the sample and which physics process it represents;
- sample name;
- whether or not the samples should be enabled by default;
- list of DBS names;
- the list of DBS names in each sample set is split by MiniAOD production campaign (eg
RunIISummer16MiniAODv3,RunIIFall17MiniAODv2) and may contain multiple DBS names per campaign in case there are extended samples available. If that's the case, the extended samples must be assigned a different sample name than what was specified for the "sample set" (because the sample names have to be as unique as the DBS names).
Here is an excerpt from the JSON file for ttH analysis:
[
{ ... },
{
"category": "Rares",
"comment": "",
"samples": [
{ ... },
{
"name": "TTTT",
"enabled": 1,
"use_case": "irreducible background",
"process": "ttbar+xx",
"datasets": {
"RunIISummer16MiniAODv2": [
{
"dbs": "/TTTT_TuneCUETP8M1_13TeV-amcatnlo-pythia8/RunIISummer16MiniAODv2-PUMoriond17_80X_mcRun2_asymptotic_2016_TrancheIV_v6-v1/MINIAODSIM"
}
],
"RunIISummer16MiniAODv3": [
{
"dbs": "/TTTT_TuneCUETP8M1_13TeV-amcatnlo-pythia8/RunIISummer16MiniAODv3-PUMoriond17_94X_mcRun2_asymptotic_v3-v2/MINIAODSIM"
}
],
"RunIIFall17MiniAOD": [
{
"dbs": "/TTTT_TuneCP5_13TeV-amcatnlo-pythia8/RunIIFall17MiniAOD-PU2017_94X_mc2017_realistic_v11-v1/MINIAODSIM"
}
],
"RunIIFall17MiniAODv2": [
{
"dbs": "/TTTT_TuneCP5_13TeV-amcatnlo-pythia8/RunIIFall17MiniAODv2-PU2017_12Apr2018_94X_mc2017_realistic_v14-v2/MINIAODSIM",
"alt": "TTTT"
},
{
"dbs": "/TTTT_TuneCP5_PSweights_13TeV-amcatnlo-pythia8/RunIIFall17MiniAODv2-PU2017_12Apr2018_94X_mc2017_realistic_v14-v1/MINIAODSIM",
"alt": "TTTT_PSweights"
}
],
"RunIIAutumn18MiniAOD": [
{
"dbs": "/TTTT_TuneCP5_13TeV-amcatnlo-pythia8/RunIIAutumn18MiniAOD-102X_upgrade2018_realistic_v15_ext1-v2/MINIAODSIM",
"alt": "TTTT_ext1"
}
]
},
},
{ ... },
]
},
{ ... },
]These JSON files are required for every analysis (ttH, HH multilepton and HH bbWW) and separate JSON files are needed for the synchronization workflow. These files are very elaborate, but fortunately they're the only files that need to be written and maintained manually: all other text files, meta-dictionaries and sample dictionaries are generated autoamtically and do not require any human intervention.
Before getting to the creation of meta-dictionary, a set of text files need to be generated with generate_dataset_table.py which takes only one input -- the dataset JSON file:
generate_dataset_table.py -i test/datasets/json/datasets.json # ttH
generate_dataset_table.py -i test/datasets/json/datasets_hh_multilepton.json # HH multilepton
generate_dataset_table.py -i test/datasets/json/datasets_hh_bbww.json # HH bbWW
generate_dataset_table.py -i test/datasets/json/datasets_sync.json # ttH sync
generate_dataset_table.py -i test/datasets/json/datasets_hh_bbww_sync.json # HH bbWW sync
The script creates two sets of files for every MC production campaign. For instance, if the JSON file specifies samples for 5 different campaigns (as in the example JSON code above) two sets of (up to) 5 files will be created. The first set of files lists the DBS names of each sample, followed by
- whether or not the sample is enabled by default;
- sample category;
- sample name;
- cross section;
- reference for the cross section value.
As with data, these files are actually used as input to launchall_nanoaod.sh script in Ntuple production.
The files contain comments (preceded with a hashtag), including the command that generated the file.
(The DBS names for MC are also used to determine which samples have more than 100'000 luminosity blocks in their "file blocks" -- otherwise the CRAB servers refuse to accept jobs for such samples when running in EventAwareLumiBased splitting mode.
This list of buggy datasets is obtainable with count_lumis_datasets.sh script which takes the list of MC DBS names as an input.
The results are stored in $CMSSW_BASE/src/tthAnalysis/NanoAOD/test/datasets_fileblock_err.txt and is automatically read by launchall_nanoaod.sh during the CRAB submission.)
The second set of text files list the sample names that cover the same phase space. In other words, it lists all sample names that have extended samples, and the sample names of the extended samples, one set per line. If there are no such sample pairs (or triplets, quadruplets, ..) the text file is not created.
All of the above is summarized into step "1" in the figure below. The two sets of files are highlighted with blue color; meta-dictionaries are highlighted with magenta.
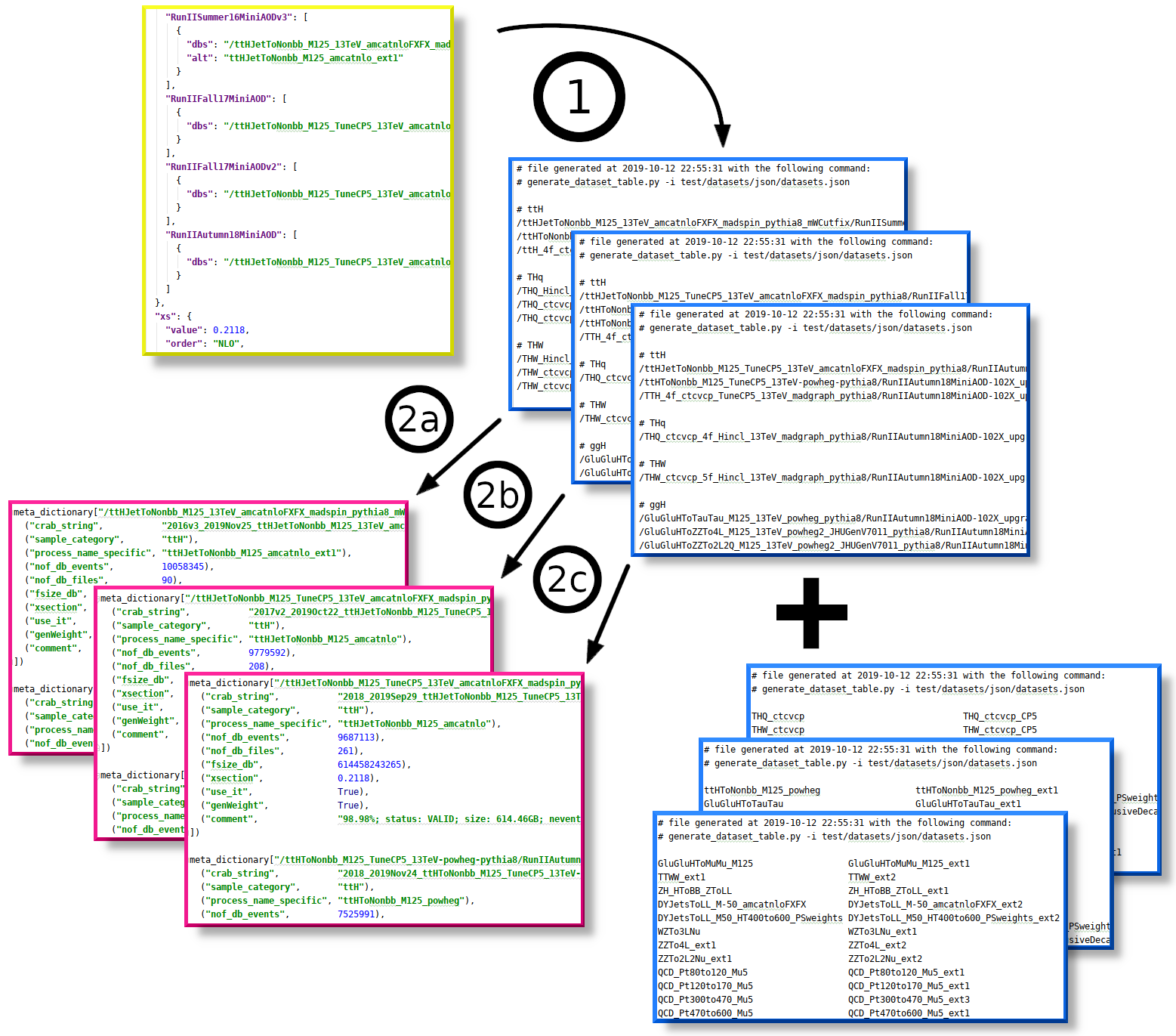
The meta-dictionary is generated by parsing the DBS names (passed via -i) and samples names that cover the same phase space (specified with -s):
# example 2a
find_samples.py -V -m python/samples/metaDict_2016_mc.py \
-i ../NanoAOD/test/datasets/txt/datasets_mc_2016_RunIISummer16MiniAODv3.txt \
-s ../NanoAOD/test/datasets/txt/sum_datasets_2016_RunIISummer16MiniAODv3.txt \
-c python/samples/sampleLocations_2016_nanoAOD.txt
# example 2b
find_samples.py -V -m python/samples/metaDict_2017_mc.py \
-i ../NanoAOD/test/datasets/txt/datasets_mc_2017_RunIIFall17MiniAODv2.txt \
-s ../NanoAOD/test/datasets/txt/sum_datasets_2017_RunIIFall17MiniAODv2.txt \
-c python/samples/sampleLocations_2017_nanoAOD.txt
# example 2c
find_samples.py -V -m python/samples/metaDict_2018_mc.py \
-i ../NanoAOD/test/datasets/txt/datasets_mc_2018_RunIIAutumn18MiniAOD.txt \
-s ../NanoAOD/test/datasets/txt/sum_datasets_2018_RunIIAutumn18MiniAOD.txt \
-c python/samples/sampleLocations_2018_nanoAOD.txtThe result is just a python dictionary that contains the same information as the meta-dictionary for data samples, except that the cross section fields are populated with proper values, and there are no fields for run ranges. Also, the list of main and extended sample names is preserved. The dictionary may contain INVALID samples -- it's the responsibility of the user to make sure that none of the samples used in the analysis are invalidated.
Sample dictionaries are needed to store
- invariant information about the dataset which is pulled from the meta-dictionaries;
- variable information about the Ntuples such as:
- number of files;
- total and average file size;
- what HLT paths should be used in the dataset;
- unweighted number of events (ie how many entries there in
TTrees in total); - LHA PDF set name (not available before post-production);
- number of LHE reweighting weights;
- acutal location of the Ntuples;
- various lists of missing HLT paths.
The sample dictionary may store information about both data and MC.
All sample dictionaries are generated with create_dictionary.py script that generally supports the following options:
-
-m-- path to the meta-dictionary (can use dictionary that combines both data and MC); -
-p-- path to sample locations (can list paths for both data and MC); -
-N-- dictionary name in the Python code; -
-E-- era; -
-o-- output directory; -
-g-- output file name; -
-M-- flag to find inconsistencies in HLT paths in a given sample.
Example:
create_dictionary.py \
-m python/samples/metaDict_2016.py \
-p python/samples/sampleLocations_2016_nanoAOD.txt \
-N samples_2016 \
-E 2016 \
-o python/samples \
-g tthAnalyzeSamples_2016_nanoAOD.py \
-MThe dictionary will be saved to python/samples/tthAnalyzeSamples_2016_nanoAOD.py.
It may take a few hours to generate the file.
Since MC Ntuples require far more time on post-production, it is preferable to post-process data and MC Ntuples seprately. The jobs for data Ntuples can take 20 input files, while the jobs for MC Ntuples shouldn't take more than 5 files as input. In practice, this can be done by adding the following to the above command:
- data:
-j 20 -f cat:data_obs; - MC:
-j 5 -f "cat:~data_obs"(tilde works as negation operator in this context).
The Ntuples are now accessible by the post-production framework. However, the post-production of MC Ntuples requires additional input: a ROOT file containing histograms for every MC sample, filled with the true number interactions per bunch crossing. These histograms are used in post-production to estimate per-event PU weight. Contents of one such file is shown in the figure below.
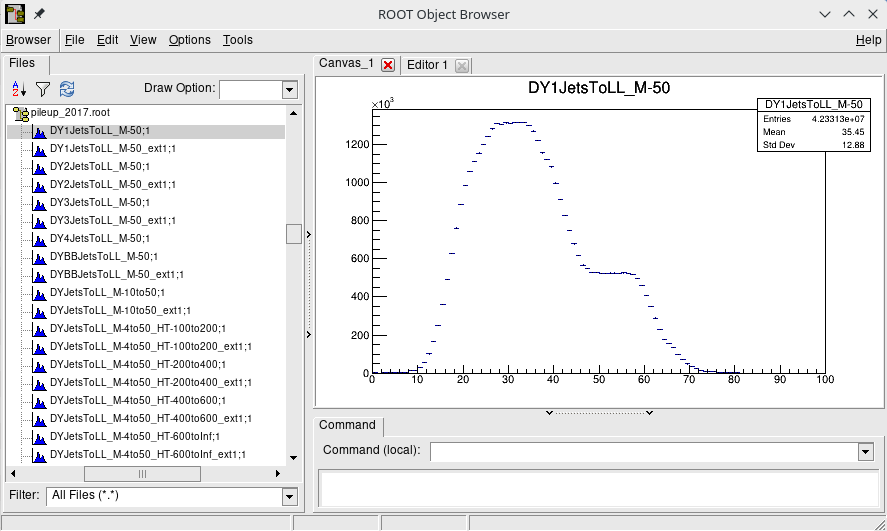
Example how to get the above file:
./test/tthProjection.py -e 2017 -v 2019Nov20 -m all -p pileup -o pileup_2017.rootIt may take up to an hour before the result will arrive on /hdfs.
The file has to be copied to the data folder in ttH repository.
Note that the b-tagging SF normalization SFs (described in this post) can be determined with the same submission script; for instance:
./test/tthProjection.py -e 2016 -v 2020Jul09 -m all -p btagSF -j 2The post-production is simply run with:
./test/tthProdNtuple.py -e $ERA -v $VERSION -m $MODEwhere $ERA has to be either 2016, 2017 or 2018, and $MODE either all (ttH data and MC Ntuples), hh (HH multilepton), hh_bbww (HH bbWW), sync (ttH sync) or hh_bbww_sync (HH bbWW sync). $VERSION can be anything.
The ttH jobs may run up to 2 days; post-production of HH datasets takes maybe half a day; time spent on post-processing sync Ntuples is in the order of hours.
This step is the same as before post-processing: only the path to the Ntuples and output file name need to be changed (note subtle differences in arguments of -p and -g options):
create_dictionary.py \
-m python/samples/metaDict_2016.py \
-p python/samples/sampleLocations_2016.txt \
-N samples_2016 \
-E 2016 \
-o python/samples \
-g tthAnalyzeSamples_2016_base.py \
-MIt shouldn't take longer than an hour for this command to finish. At this point, the Ntuples are finally usable in the analysis framework.
There's one extra step required for non-resonant HH samples: getting the so-called denominator histograms needed in HH reweighting. The denominator histogram (shown below) is 2D histogram binned in di-Higgs mass and cosine angle of the di-Higgs system at the LHE particle level.
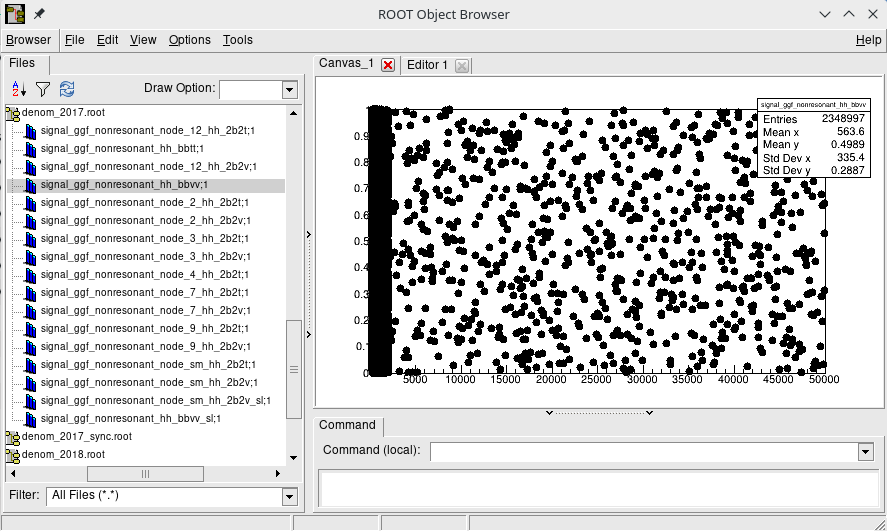
These histograms can be found by running:
./test/hhNonResDenom.py -e $ERA -v $VERSION -m $MODEin $CMSSW_BASE/src/hhAnalysis/multilepton.
Acceptable values for $MODE are: hh_multilepton, hh_bbww, hh_bbww_sync.
Similarly to PU histogram case, the output will be stored to /hdfs and must be copied to the data directory of a given repository.
The jobs may take up to an hour to run.
Although the Ntuples are now fully compatible with the analysis framework, they're simply too large to analyze with full systematics. In order to get around this problem, the Ntuples have to be skimmed, or filtered by some loose criteria that satisfies most analysis requirements.
In practice, this step is identical to the post-processing, except that the skimming is enabled with -p:
./test/tthProdNtuple.py -e $ERA -v $VERSION -m $MODE -pBecause this step throws out a lot of events, it may create lots of small files.
The solution is to use more input files per job, for instance 20 (just adding -j 20 will do the trick).
Note that there's really no need to run skimming separately for data and MC, because the the time-consuming part (adding more information to the Ntuples) is not enabled.
The skimming shouldn't take more than few hours to complete.
The sample dictionary for the skimmed samples can be generated with eg:
create_dictionary.py \
-m python/samples/metaDict_2016.py \
-p /hdfs/local/karl/ttHNtupleProduction/2016/2019Dec12_wPresel_nom_all/ntuples \
-N samples_2016 \
-E 2016 \
-o python/samples \
-g tthAnalyzeSamples_2016_preselected_base.py \
-MNotice that the sample location parameter -p also accepts the actual paths to the Ntuples.
At this point, the Ntuples are ready for all analyses, with the exception of MEM.
Some analyses, such as 2lss+1tau and 3l in ttH, may require MEM computation. Because the MEM computation is very CPU-intense, it is desirable to compute the scores only once per event. It follows that the MEM information should be added to the existing Ntuples and read back at the analysis level, instead of computing the MEM score on the fly in the analysis.
However, computing the MEM scores for all events is also unfeasible due to sheer number of events in each era. The workaround is to determine the list of events that enter the signal region or fake application region of a given channel that implements MEM. Systematic shifts of observables that affect the event selection (tauES, JES, JER) must also be taken into account.
The idea is to run a given analysis with full systematics, for example:
./test/tthAnalyzeRun_2lss_1tau.py -e $ERA -v $VERSION -m default -s full -GThen obtain the list of run-lumi-event numbers for each sample and analysis region, and construct a lookup table with:
inspect_rle_numbers.py \
-i /hdfs/local/$USER/ttHAnalysis/$ERA/$VERSION \
-o data/mem/mem_${ERA}_deepVSjVLoose.root \
-w 2lss_1tauThis lookup table (depicted below) is used as a filter in MEM workflow: if the event is not present in the lookup table, the MEM computation is skipped for the event. This ensures that MEM scores are found only for the events that actually enter the analysis.
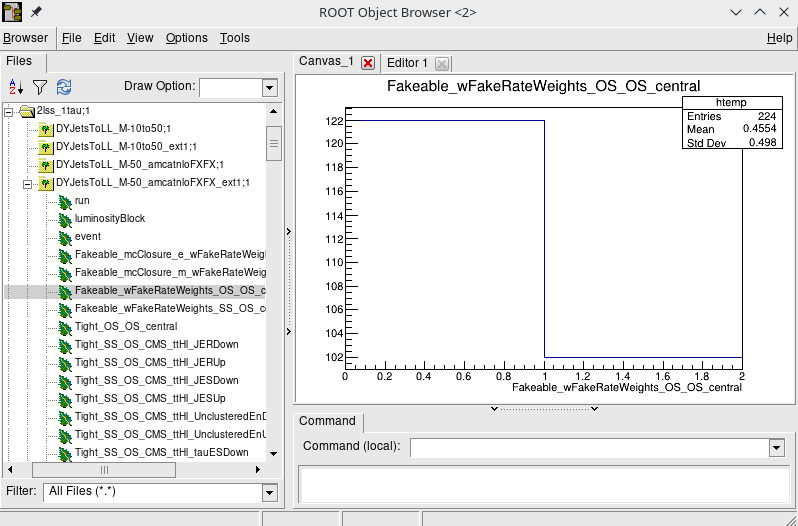
The analysis jobs may take a day to complete, but constructing the so-called filter file is a matter of minutes.
The filter file created in the previous step is automatically read during the creation of configuration files to the MEM jobs:
./test/addMEM_2lss_1tau.py -e $ERA -v $VERSION -m defaultEstimates vary, but these jobs may take a week or two to finish.
The Ntuples with MEM scores attached become usable in the analysis framework once there is a sample dictionary available to access them:
create_dictionary.py \
-m python/samples/metaDict_${ERA}.py \
-p /hdfs/local/$USER/addMEM/$ERA/${VERSION}_default_nom_high/final_ntuples/2lss_1tau \
-N samples_${ERA} \
-E $ERA \
-o python/samples \
-g tthAnalyzeSamples_${ERA}_addMEM_2lss1tau_deepVSjVLoose.py \
-M