Transitions - Genometric/GeMSE GitHub Wiki
GeMSE supports the following transitions: Extract, Sort, Rewrite, Discretize, and Clustering.
Follow these steps to apply a transition:

- From start-transition tree, choose a node on which the transition should be applied.
- Choose a transition from
Operationsdropdown. - (Optional) assign a label for the transition in the
Operation Labeltextbox. - Set up the transition-specific parameters (described in the following for each transition).
- Click the
Apply Operationbutton.
The transitions and their parameters are described in the following.
Extracts a sub-genometric space from a genometric space.
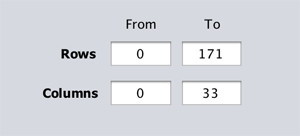
This transition has the following arguments:
-
Rows-From(inclusive): sets the row number from which the extraction starts. -
Rows-To(exclusive): sets the row number at which the extraction ends. -
Columns-From(inclusive): sets the column number from which the extraction starts. -
Columns-To(exclusive): sets the column number at which the extraction ends.
Sorts in Ascending or Descending order the selected genometric space based on various attributes.
Sorting Area is the domain of the attributes for a sort criterion; hence, Metadata and Sorting domain are options that vary depending on the Sorting Area option. The Sorting Areas are as follows:
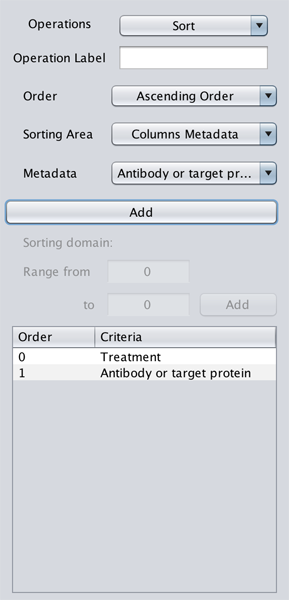
- Columns Metadata: sorts columns based on the metadata of the loaded samples. When this option is selected, GeMSE populates the
Metadatadropdown with all the loaded metadata attributes. User chooses an option, then clicks on theAddbutton to add the criterion. Multiple attributes can be defined, and columns will be sorted based on the attributes in their entered order. - Rows Metadata: sorts rows based on a
stringattribute of the loaded samples (e.g., gene name). - Columns Contents: sorts columns based on the values in the cells. User defines a range of columns whose cells should be used for sorting, using the
Range FromandToarguments. If theToargument is set more than the number of columns, GeMSE automatically adds only the available column having clicked theAddbutton. - Rows Contents: sorts rows based on the values defined in the cells. Parameter setting is similar to
Columns Contents.
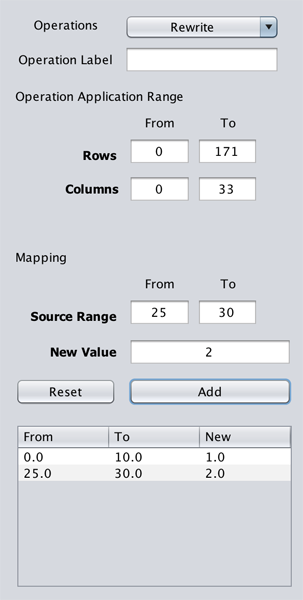
The Rewrite transition is applicable on the whole, or a sub-genometric space, of the selected genometric space. By default, the Rows-From/To and Columns-From/To are set to target the selected genometric space entirely.
The Source Range parameter with From and To arguments, sets a range of values to be rewritten with the value given for the New Value parameter.
After setting the parameters, clicking on Add button registers the mapping (not applied yet).
User can define an arbitrary set of mappings. All the registered mappings are displayed in the table below the Add button. The registered mappings can be deleted clicking on Reset button. The mappings are applied having clicked on the Apply Operation button.
Note that, the defined mappings are discreet (see Discretize for contiguous rewrite). In other words:
- the
Rewriteoperation do not necessarily rewrite all the values in the selected sub-genometric space (the values which are not covered by the mappings do not change) - the defined mappings can overlap, and the values are changed to the last mapping in a group of overlapping mappings.
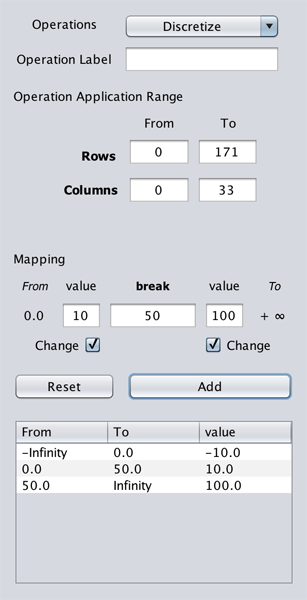
Unlike Rewrite, Discretize is a contiguous rewrite. Similar to Rewrite, Discretize can be applied on a sub-genometric space of a selected genometric space. The operation is setup as following.
Choose a break point. This point divides the values of the selected genometric space in two groups:
- the values less than or equal to the
breakand greater than a previously defined smallerbreak. All the values in this range are replaced with thevalueentered in the textbox to the left of thebreaktextbox. Or, not changed ifChangeis unchecked. - the values greater than the
breakand less than a previously defined greaterbreak. All the values in this range are replaced with thevalueentered in the textbox to the right of thebreaktextbox. Or, not changed ifChangeis unchecked.
For instance:
-
Set the
breakto0, and enter-10and10in the left and rightvaluetextboxes respectively. This defines a mapping as:(-∞, 0] ← -10
(0, ∞) ← 10
-
Then set the
breakto50and enter10and100in the left and rightvaluetextboxes respectively. This updates the mapping as follows:(-∞, 0] ← -10
(0, 50] ← 10
(50, ∞) ← 100
GeMSE automatically updates the From and To labels based on the entered break and previously defined mappings.
The mapping is registered by clicking on the Add button. All the defined mappings are listed in the mappings table.
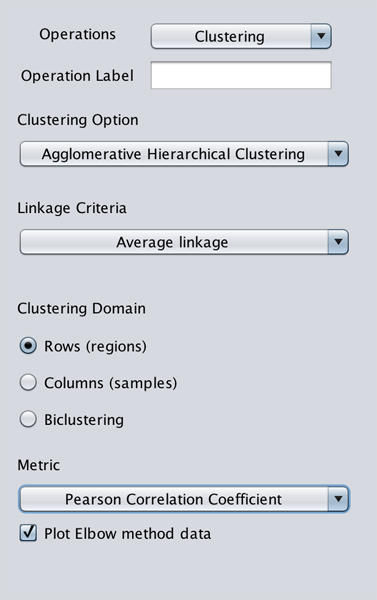
This operation clusters data using Agglomerative Hierarchical Clustering. The linkage criteria options are:
And clustering can be applied on rows, columns, or both (aka bi-clustering; it requires a connection to R to be set a prior, see setup page). The clustering metrics are:
- Euclidean Distance
- Manhattan Distance
- Earth Movers Distance
- Chebyshev Distance
- Canberra Distance
- Pearson Correlation Coefficient
Additionally, GeMSE suggests a number of clusters using the Elbow method. If Plot Elbow method data is checked, it plots percentage of variation vs number of clusters, and suggests a number of clusters based on highest percentage of variation slope change.