Load Data - Genometric/GeMSE GitHub Wiki
GeMSE operates on genomic features present in an interval-based format, such as ChIP-seq peaks. GeMSE takes its input in data (e.g., see this file), and metadata (e.g., see this file). Data and metadata are two files in the same directory with the metadata file having the same name as the data filename with a .meta postfix. For instance, the data file DEX_005nM_01h_GR.bed has DEX_005nM_01h_GR.bed.meta metadata file. The files are discussed in the following.
The metadata files provide explanatory information of the data. The metadata files are optional, and they are automatically loaded if present (if they follow the aforementioned naming convention); if not present, data still will be loaded but without explanatory information.
The metadata is a plain text file, where each line in the file is an attribute-value pair. For instance:
Treatment DEX_5nM 1h
Antibody or target protein GR (sc-1003)
where Treatment and Antibody or target protein are the attributes, and respectively DEX_5nM 1h and GR (sc-1003) are their values. See this file as a complete example.
A user does not need to specify a metadata file, or load it separately. If a metadata file is available and following the aforementioned naming convention, then GeMSE loads the metadata files automatically when loading data files.
The data is a set of intervals each abstracting a characteristic of a region on the genome (e.g., ChIP-seq peaks in BED files, or mutation in VCF files). GeMSE loads data from tab-delimited files such as BED, ENCODE broadPeak and narrowPeak, GTF, or an arbitrary CSV file. For instance, see this file. The data files can be generally grouped in two categories: BED-like and GTF. GeMSE reads three required columns chromosome, start, and stop. However, depending on the file type GeMSE reads various values; discussed as follows.
For such file types, GeMSE requires at least one numerical column. When such file types are selected, GeMSE shows the following window.
 |
|---|
| Figure 1. |
- Section 1 shows the first few lines (15 lines) of the selected sample.
- Section 2 provides an option of specifying the number of header lines which should not be read.
-
Section 3 provides features for the user to configure the parser on reading any of the numerical columns--parser requires the
chromosome,start, andstopto be respectively the first, second, and the third columns. In the shown example, the forth column contains names of the p-values; which is not a numeric attribute. To avoid this column, the checkboxValue is numeric, and read the columnshould be unchecked. The buttonsPrevious ColumnandNext Columnswitches between columns. In this example,Next Columnallows configuring the parser for the fifth column, which is theX-squared. This column has the numeric attribute of the regions, hence it should be read by checking theValue is numeric, and read the columncheckbox. Assigning a label to each column is optional. However, since this label is used in GeMSE to refer to the data read from the column, it is recommended to provide a short explanatory label.
The settings are used for loading all the selected samples. Samples in different formats are loaded separately. Having configured the settings, clicking on OK button GeMSE caches the selected samples. Then it reports the results in a window as the following.
 |
|---|
| Figure 2. |
This window provides an overview of Loaded Samples, and any error messages in the Details tab. The Load Samples tab shows the number of lines in each sample, and the number of loaded lines (Feature count). If the numbers in the two columns are different, then the line number of the dropped line and a reason for dropping it is available in the Details tab. See the following figure.
 |
|---|
| Figure 3. |
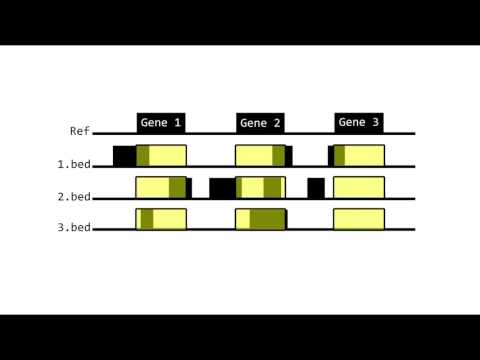
For instance, line #1 of sample 1.bed is dropped for two reasons: invalid start\stop column values, and invalid Column 5, which is expected to be x-squared (a numeric value) while it is X-squared (a string value). The line #1 is a header line. Since the number of header lines on Section 2 was set to 0, GeMSE attempted parsing it. However, since the _line #1' does not adhere to the defined requirements, GeMSE dropped the line. Accordingly, any line in the sample (at any part of it) which do not adhere to the requirements (e.g., the required chromosome, start, and stop columns) and the user-defined parameters, will not be read. The ignored lines, and the reason for ignoring them is reported.
The temporarily cached samples are loaded in GeMSE by clicking on the YES button.
GeMSE reads GTF files in standard format, which is:
| Column number | Attribute | Read | Description |
|---|---|---|---|
| 1 | chromosome | ✔ | with or without chr prefix |
| 2 | source | ✘ | data source |
| 3 | feature | ✔ | feature type name, e.g. Gene |
| 4 | start | ✔ | start position of the feature |
| 5 | stop | ✔ | end position of the feature |
| 6 | score | ✘ | a floating point value |
| 7 | strand | ✘ | defined as + (forward) or - (reverse) |
| 8 | frame | ✘ |
0, 1 or 2 indicates the base of the feature which is the first base of a codon |
| 9 | attribute | ✔ | a semicolon-separated list of attribute-value pairs |
(see the Ensembl page for the GTF format details)
Attributes with numeric values at column #9, are used as values for GenoMetric Space cells, and attributes with string values are used for labeling and descriptive information in pattern extraction section.
Once the selected GTF files are loaded, the results are displayed on a windows as shown on Figure 2.
A prerequisite in GeMSE is: all the loaded files refer to common positions on the genome with different values. Therefore, all the files have same number of feature, with same chromosome, start and stop attributes, and in the same order. GeMSE refers to such files as homogeneous files, and the files that do not meet this requirement are considered heterogeneous files.
When loading samples, GeMSE checks for the heterogeneity of the files. If homogeneous, then it loads the cached files. If heterogeneous, then it shows the following window.
 |
|---|
| Figure 4. |
Using a reference sample (e.g., Ensemble reference genes for hg19
assembly), GeMSE can convert heterogeneous files to homogeneous files. Clicking on OK on the above window, GeMSE shows the following window.
 |
|---|
| Figure 5. |
Select a reference sample using the Browse button. Then select an attribute of the heterogeneous files from the Attribute to aggregate dropdown, and specify an Aggregation function. Clicking on Load button, GeMSE first loads the reference sample, then it converts the heterogeneous samples to homogeneous samples, then it shows the load results on a window as shown on Figure 2.