5. Feature Table Construction - DianaCarolinaVergara/16S-rRNA-Analysis GitHub Wiki
Feature Table
With this new version of Qiime, we call this an feature table (OTUs table with Qiime1).
OTUs (Operational Taxonomic Units) are sequences selected from the reads. An operational taxonomic unit (OTU) is an operational definition used to classify groups of closely related individuals. The term was originally introduced by Robert R. Sokal and Peter H. A. Sneath in the context of numerical taxonomy, where an "Operational Taxonomic Unit" is simply the group of organisms currently being studied*.
The goal is to identify a set of correct biological sequences.
An OTU table is a matrix that gives the number of reads per sample per OTU. One entry in the table is usually a number of reads, also called a "count", or a frequency in the range 0.0 to 1.0. Counts are calculated by mapping reads to OTUs. Note that read abundance has very low correlation with species abundance, so the biological interpretation of these counts is unclear. I suggest making OTU tables for 97% OTUs.
Qiime classic OTU table:
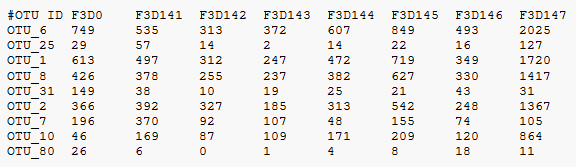
First, we need the command qiime feature-table summarize and some files to do the feature table. We need the previously generated quality filtered dada2-table table-dada2, and a metadata file containing all the important information of the saamples collected:
SampleID, reads, center_name, instrument_model, library_construction_protocol, platform, run_center_ target_gene, target_subfragment.
In my specific case as I work with coral, I include some biological ang geographyc information:
genus, species, country, locality, depth, date, zooxanthellae (AZ-Z), etc.
e.g.

Code:
qiime feature-table summarize \
--i-table table-dada2.qza \
--m-sample-metadata-file sample_metadata.txt \
--o-visualization full-table.qzv
Here we obtained the first .qzv file.
To open this file we use the qiime view platform. You just have to take the .qzv file and 'drag and drop' in that page.

We have to delete the repetitive seqs.
qiime feature-table tabulate-seqs \
--i-data rep-seqs-dada2.qza \
--o-visualization rep-seqs.qzv
qiime fragment-insertion sepp \
--i-representative-sequences rep-seqs-dada2.qza \
--p-threads 4 \
--o-tree insertion-tree.qza \
--o-placements insertion-placements.qza
Summarize and convert the -.qza in a visualization file -.qzv
qiime feature-table summarize \
--i-table table-dada2.qza \
--m-sample-metadata-file sample_metadata.txt \
--o-visualization table.qzv
And generate a summarized file with the removed reads removed-table.qzv.
qiime feature-table summarize \
--i-table table-dada2.qza \
--m-sample-metadata-file sample_metadata.txt \
--o-visualization removed-table.qzv
References
-
Sokal & Sneath: Principles of Numerical Taxonomy, San Francisco: W.H. Freeman, 1963
-
Blaxter, M.; Mann, J.; Chapman, T.; Thomas, F.; Whitton, C.; Floyd, R.; Abebe, E. (October 2005). "Defining operational taxonomic units using DNA barcode data". Philos Trans R Soc Lond B Biol Sci. 360 (1462): 1935–43.