Automated Visualization of mincLm and mincLmer Models - CoBrALab/documentation GitHub Wiki
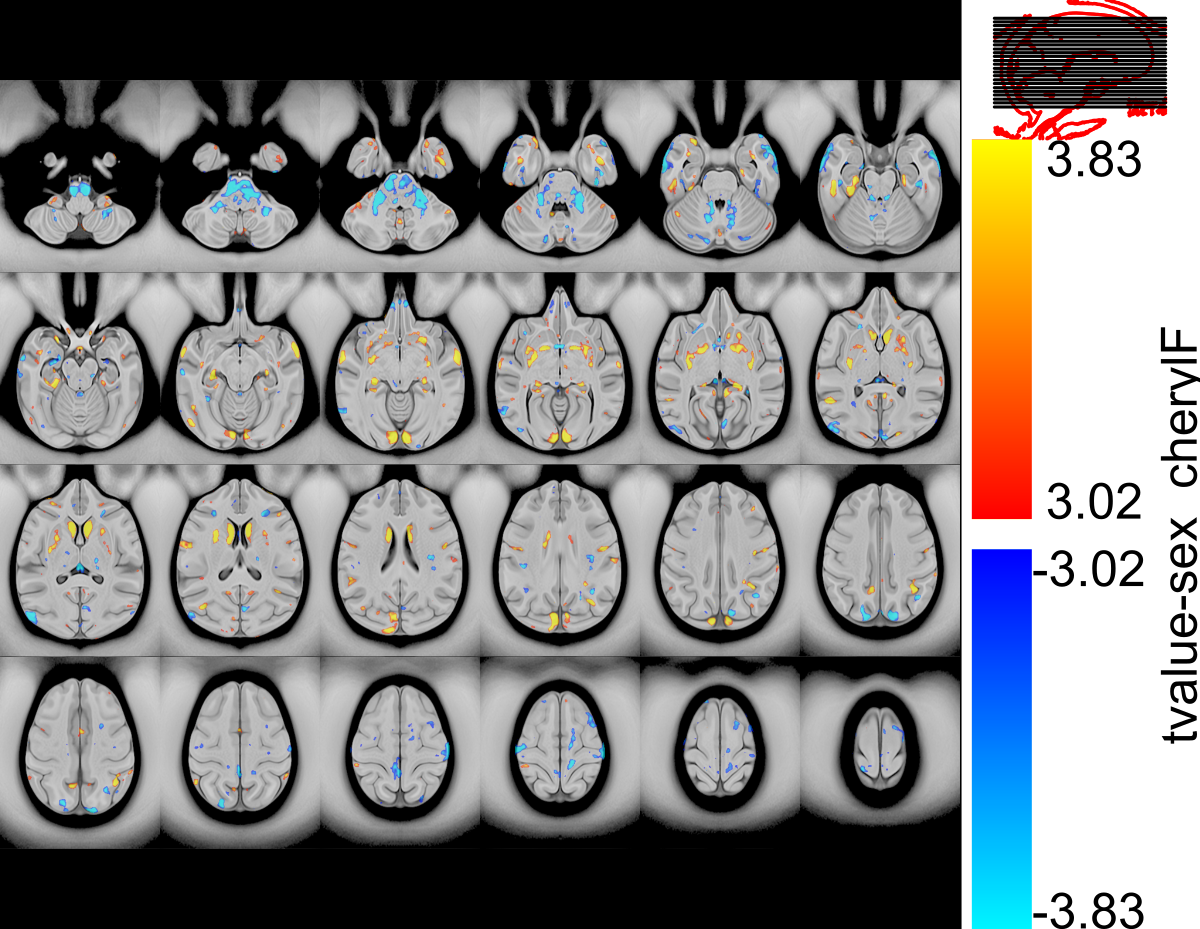
Automatic Visualization with transparent thresholds
This visualization is built on the recommendations of:
Go Figure: Transparency in neuroscience images preserves context and clarifies interpretation https://arxiv.org/abs/2504.07824#
In this figure, the significance map below the 0.05 threshold is faded out in a linear fasion to t-value 0. Highlighting contours of black (5% FDR threshold) show exactly where regions of significance begin.
library(grid)
library(tidyverse)
library(MRIcrotome)
library(RMINC)
library(magrittr) #to be able to use "%>%"
## HERE GOES CODE TO SETUP DATAFRAME ETC ##
anatVol <- mincArray(mincGetVolume("template.mnc"))
averagemask <- mincArray(mincGetVolume("ask.mnc"))
model = mincLmer(relative ~ poly(age,2)*sex + (1|subject) + (1|dataset), data = data, mask = "mask.mnc", parallel = c("local",20))
model = mincLmerEstimateDF(model)
thresholds = attr(mincFDR(model), "thresholds")
print(thresholds)
#Here we figure out the extent of the mask, so we can use it to limit
# the FOV
# You may want to adjust these slighty buy adding/subtracting, so you don't
# quite go to the edge of the mask
dim1_begin <- min(which(averagemask == 1, arr.ind=TRUE)[,"dim1"])
dim1_end <-max(which(averagemask == 1, arr.ind=TRUE)[,"dim1"])
dim2_begin <- min(which(averagemask == 1, arr.ind=TRUE)[,"dim2"])
dim2_end <- max(which(averagemask == 1, arr.ind=TRUE)[,"dim2"])
dim3_begin <- min(which(averagemask == 1, arr.ind=TRUE)[,"dim3"])
dim3_end <- max(which(averagemask == 1, arr.ind=TRUE)[,"dim3"])
jacobian="relative"
# If your model is instead a mincLm, you need to use this loop command
# #for (predictor in dimnames(thresholds)[2](/CoBrALab/documentation/wiki/2)[c(-1,-2)]) {
for (predictor in dimnames(thresholds)[2](/CoBrALab/documentation/wiki/2)[c(-1)]) {
#If you want to save this to a file, uncomment next line
#svg(paste0("results/human_",jacobian,"_",predictor,".svg"), height = 3.1, width = 4)
#We use a tryCatch here to keep going if there's some kind of error in a an individual plot
tryCatch({
#Here, we extract the thresholds from the code, and clip them to 2 digits, otherwise the plotting doesn't look good
lowerthreshold = round(thresholds["0.05",predictor],digits=2)
#Sometimes, there isn't an 0.01 threshold, when thats the case, we use the max instead, be careful to read the threshold array printed above
upperthreshold = round(ifelse(is.na(thresholds["0.01",predictor]), max(c(max(mincArray(model, predictor)),abs(min(mincArray(model, predictor))))), thresholds["0.01",predictor]),digits=2)
# Generate the default colourmaps
pospal = colorRampPalette(c("red", "yellow"), alpha=TRUE)(255)
negpal = colorRampPalette(c("blue", "turquoise1"), alpha=TRUE)(255)
# Find the crossover point in the map where the colourmap switches to "non transparent"
# Need to include the alpha term here, but its always opaque
breakpointpos = pospal[round(lowerthreshold/upperthreshold*255) - 1]
breakpointneg = negpal[round(lowerthreshold/upperthreshold*255) - 1]
# Generate a subset of the colourmap now which ramps from the same starting point and ends at breakpoint, with full opacity
pospalalpha = colorRampPalette(c("#FF000000", breakpointpos), alpha=TRUE)(round(lowerthreshold/upperthreshold*255) - 1)
negpalalpha = colorRampPalette(c("#0000FF00", breakpointneg), alpha=TRUE)(round(lowerthreshold/upperthreshold*255) - 1)
# Concatenate the two maps together for a complete map
combinedpospal = c(pospalalpha, pospal[round(lowerthreshold/upperthreshold*255):length(pospal)])
combinednegpal = c(negpalalpha, negpal[round(lowerthreshold/upperthreshold*255):length(negpal)])
# Here is the plotting code, we do all three slice directions in one figure. This was optimized for a human brain, you may need to
# adjust the nrow and ncol to get exactly the figure you want
# You will also need to adjust the anatVol low and high values to correspond to good thresholds for your template
sliceSeries(nrow = 5, ncol = 2, dimension = 2, begin = dim2_begin, end = dim2_end) %>%
anatomy(anatVol, low=1, high=5.9) %>%
addtitle("Coronal") %>%
overlay(mincArray(model, predictor),
low = 0,
high = upperthreshold,
col = combinedpospal,
rCol = combinednegpal,
symmetric = T) %>%
contours(abs(mincArray(model, predictor)), levels=lowerthreshold, lwd=2, col="black") %>%
sliceSeries(nrow = 6, ncol= 2, dimension = 1, begin = dim1_begin, end = dim1_end) %>%
anatomy(anatVol, low=1, high=5.9) %>%
addtitle("Sagittal") %>%
overlay(mincArray(model, predictor),
low = 0,
high = upperthreshold,
col = combinedpospal,
rCol = combinednegpal,
symmetric = T) %>%
contours(abs(mincArray(model, predictor)), levels=lowerthreshold, lwd=2, col="black") %>%
sliceSeries(nrow = 5, ncol= 2, dimension = 3, begin = dim3_begin, end = dim3_end) %>%
anatomy(anatVol, low=1, high=5.9) %>%
addtitle("Axial") %>%
overlay(mincArray(model, predictor),
low = 0,
high = upperthreshold,
col = combinedpospal,
rCol = combinednegpal,
symmetric = T) %>%
legend(predictor) %>%
contours(abs(mincArray(model, predictor)), levels=lowerthreshold, lwd=2, col="black") %>%
draw()}, error=function(e){cat("ERROR :",conditionMessage(e), "\n")})
#If you are saving to file, also uncomment this
#dev.off()
Cropping stats/model to improve visualization
Here we use the existing averagemask already provided in the data to crop the template and the stats output before visualization
# Set required padding around mask in voxels (adjust as needed
new_padding <- 5
# Get new bounds
bounds <- which(averagemask > 0.5, arr.ind = T) %>%
as_tibble() %>%
gather(dim, index) %>%
group_by(dim) %>%
summarize(
min_slice=(min(index) - new_padding),
max_slice=(max(index) + new_padding)
)
anatVol_cropped <- anatVol[bounds$min_slice[1](/CoBrALab/documentation/wiki/1):bounds$max_slice[1](/CoBrALab/documentation/wiki/1),
bounds$min_slice[2](/CoBrALab/documentation/wiki/2):bounds$max_slice[2](/CoBrALab/documentation/wiki/2),
bounds$min_slice[3](/CoBrALab/documentation/wiki/3):bounds$max_slice[3](/CoBrALab/documentation/wiki/3)]
# This needs to go inside the visualization loop
statsarray_cropped <- mincArray(model, predictor)[bounds$min_slice[1](/CoBrALab/documentation/wiki/1):bounds$max_slice[1](/CoBrALab/documentation/wiki/1),
bounds$min_slice[2](/CoBrALab/documentation/wiki/2):bounds$max_slice[2](/CoBrALab/documentation/wiki/2),
bounds$min_slice[3](/CoBrALab/documentation/wiki/3):bounds$max_slice[3](/CoBrALab/documentation/wiki/3)]
# Now replace anatVol with anatVol_cropped and mincArray(model, predictor) with statsarray_cropped in visualization commands
# You will also need to adjust or remove the begin/end values as those refer to the original images
Classic Thresholding
Here you can find some example code which produces automatic figures from a model, in the below example, the code will produce a unified figure for each predictor, showing a triplanar view:

Here, we assume you have a dataframe data which contains all your information.
library(grid)
library(tidyverse)
library(MRIcrotome)
library(RMINC)
library(magrittr) #to be able to use "%>%"
## HERE GOES CODE TO SETUP DATAFRAME ETC ##
anatVol <- mincArray(mincGetVolume("template.mnc"))
averagemask <- mincArray(mincGetVolume("ask.mnc"))
model = mincLmer(relative ~ poly(age,2)*sex + (1|subject) + (1|dataset), data = data, mask = "mask.mnc", parallel = c("local",20))
model = mincLmerEstimateDF(model)
thresholds = attr(mincFDR(model), "thresholds")
print(thresholds)
#Here we figure out the extent of the mask, so we can use it to limit
# the FOV
# You may want to adjust these slighty buy adding/subtracting, so you don't
# quite go to the edge of the mask
dim1_begin <- min(which(averagemask == 1, arr.ind=TRUE)[,"dim1"])
dim1_end <-max(which(averagemask == 1, arr.ind=TRUE)[,"dim1"])
dim2_begin <- min(which(averagemask == 1, arr.ind=TRUE)[,"dim2"])
dim2_end <- max(which(averagemask == 1, arr.ind=TRUE)[,"dim2"])
dim3_begin <- min(which(averagemask == 1, arr.ind=TRUE)[,"dim3"])
dim3_end <- max(which(averagemask == 1, arr.ind=TRUE)[,"dim3"])
jacobian="relative"
# If your model is instead a mincLm, you need to use this loop command
# #for (predictor in dimnames(thresholds)[2](/CoBrALab/documentation/wiki/2)[c(-1,-2)]) {
for (predictor in dimnames(thresholds)[2](/CoBrALab/documentation/wiki/2)[c(-1)]) {
#If you want to save this to a file, uncomment next line
#svg(paste0("results/human_",jacobian,"_",predictor,".svg"), height = 3.1, width = 4)
#We use a tryCatch here to keep going if there's some kind of error in a an individual plot
tryCatch({
#Here, we extract the thresholds from the code, and clip them to 2 digits, otherwise the plotting doesn't look good
lowerthreshold = round(thresholds["0.05",predictor],digits=2)
#Sometimes, there isn't an 0.01 threshold, when thats the case, we use the max instead, be careful to read the threshold array printed above
upperthreshold = round(ifelse(is.na(thresholds["0.01",predictor]), max(c(max(mincArray(model, predictor)),abs(min(mincArray(model, predictor))))), thresholds["0.01",predictor]),digits=2)
# Here is the plotting code, we do all three slice directions in one figure. This was optimized for a human brain, you may need to
# adjust the nrow and ncol to get exactly the figure you want
# You will also need to adjust the anatVol low and high values to correspond to good thresholds for your template
sliceSeries(nrow = 5, ncol = 2, dimension = 2, begin = dim2_begin, end = dim2_end) %>%
anatomy(anatVol, low=1, high=5.9) %>%
addtitle("Coronal") %>%
overlay(mincArray(model, predictor),
low=lowerthreshold,
high=upperthreshold,
symmetric = T, alpha=0.6) %>%
sliceSeries(nrow = 6, ncol= 2, dimension = 1, begin = dim1_begin, end = dim1_end) %>%
anatomy(anatVol, low=1, high=5.9) %>%
addtitle("Sagittal") %>%
overlay(mincArray(model, predictor),
low=lowerthreshold,
high=upperthreshold,
symmetric = T, alpha=0.6) %>%
sliceSeries(nrow = 5, ncol= 2, dimension = 3, begin = dim3_begin, end = dim3_end) %>%
anatomy(anatVol, low=1, high=5.9) %>%
addtitle("Axial") %>%
overlay(mincArray(model, predictor),
low=lowerthreshold,
high=upperthreshold,
symmetric = T, alpha=0.6) %>%
legend(predictor) %>%
draw()}, error=function(e){cat("ERROR :",conditionMessage(e), "\n")})
#If you are saving to file, also uncomment this
#dev.off()
}
Multiple figures
Here, we adjust the code to generate three figures, one for each view:
jacobian="relative"
for (predictor in dimnames(thresholds)[2](/CoBrALab/documentation/wiki/2)[-1]) {
#We use a tryCatch here to keep going if there's some kind of error in a an individual plot
tryCatch({
#Here, we extract the thresholds from the code, and clip them to 2 digits, otherwise the plotting doesn't look good
lowerthreshold = round(thresholds["0.05",predictor],digits=2)
#Sometimes, there isn't an 0.01 threshold, when thats the case, we use the max instead, be careful to read the threshold array printed above
upperthreshold = round(ifelse(is.na(thresholds["0.01",predictor]), max(c(max(mincArray(model, predictor)),abs(min(mincArray(model, predictor))))), thresholds["0.01",predictor]),digits=2)
#svg(paste0("results/",jacobian,"_",predictor,"_coronal.svg"), height = 3.1, width = 4)
sliceSeries(nrow = 5, ncol= 6, begin = dim2_begin, end = dim2_end, dimension = 2) %>% #slice sequence to display stats
anatomy(anatVol, low=1, high=5.9) %>%
overlay(mincArray(model, predictor), # specify which column of the mincLM model
low=lowerthreshold, high=upperthreshold, symmetric = T, alpha=0.6) %>% # t score range; symmetric = T means show positive and negative t values
legend(predictor) %>%
contourSliceIndicator(anatVol, c(1.8, 2), slice=125) %>%
draw()
#dev.off()
#svg(paste0("results/",jacobian,"_",predictor,"_sagittal.svg"), height = 3.1, width = 4)
sliceSeries(nrow = 5, ncol= 5, begin = dim1_begin, end = dim1_end, dimension = 1) %>% #slice sequence to display stats
anatomy(anatVol, low=1, high=5.9) %>%
overlay(mincArray(model, predictor), # specify which column of the mincLM model
low=lowerthreshold, high=upperthreshold, symmetric = T, alpha=0.6) %>% # t score range; symmetric = T means show positive and negative t values
contourSliceIndicator(anatVol, c(1.8, 2), slice=125) %>%
legend(predictor) %>%
draw()
#dev.off()
#svg(paste0("results/",jacobian,"_",predictor,"_axial.svg"), height = 3.1, width = 4)
sliceSeries(nrow = 4, ncol= 6, begin = dim3_begin, end = dim3_end, dimension = 3) %>% #slice sequence to display stats
anatomy(anatVol, low=1, high=5.9) %>%
overlay(mincArray(model, predictor), # specify which column of the mincLM model
low=lowerthreshold, high=upperthreshold, symmetric = T, alpha=0.6) %>% # t score range; symmetric = T means show positive and negative t values
legend(predictor) %>%
contourSliceIndicator(anatVol, c(1.8, 2), slice=125) %>%
draw()
#dev.off()
}, error=function(e){cat("ERROR :",conditionMessage(e), "\n")})
}