November 2021 - Bozhie/transcription-modeling GitHub Wiki
11.02.2021
Using mm9 RefSeq for TSS analysis
Purpose: Redoing analysis using actual TSS
Process/Notes:
- Using the RefSeq GTF files downloaded from UCSC browser, find TSS
- Simply took the first index of all the transcripts in the file, depending on the strand/orientation
- Repeating scatter-plot and mapping by index method from last time, looks like a tighter relationship/much less outliers
- Also noticed that using bioframe to find closest overlap between transcripts and TSS, >90% of them had a distance of 0 (makes sense)
images/20211102_TSS_v_CTCF_scatter_redo.png
{kind=link}
- TSS binning with pybbi
- Should be able to just use the TSS windows build from
- If they're from the same genome, then the stackup should filter according to the windows (I think this means we don't need to match / filter back from TSS)
- ISSUE: Still received error
ValueError: Start exceeds the chromosome length, 15902555.
11.03.2021
Explorations of sleuth-Kalliso
Resources
(Bray 2016) Near-Optimal probabilistic RNA-seq Quantification sleuth walkthrough Kallisto getting started
Notes
- Starting new directory in pokorny/RNAseq
- Goal: follow the kallisto tutorial, using RNA-seq data from Rao 2017
- Added transcriptome from NCBI GRCm38 choosing the following download settings: REfSeq source database, filetype: CDS from genomic fasta, saved in
scratch/pokorny/genomic_annotations/mm10/genome_assemblies_cds_fasta.tar - SRA download from the cloud might be another way to download raw fastq results from individual experiments
- Tried to download GSE106886_RAW.tar from Rao_2017, seems to be a huge file and tar -xv but lost remote connection while waiting.
- Submitting unzip as a job (id: Submitted batch job 6543562)
- Added transcriptome from NCBI GRCm38 choosing the following download settings: REfSeq source database, filetype: CDS from genomic fasta, saved in
Meeting with Geoff
Goals for EOW:
- Check if cleaning up TSS windows by removing anything with '_random' chrom location helps
- Continue trying to run kallisto/sleuth -- minimally, get a good RNAseq dataset downloaded
- Rao_2017 nascient PROseq data might not be the best/easiest dataset to work with here
- Where is raw RNAseq from Elphege et al?
Notes:
- Look into SRA downloads to retrieve RNAseq data. Also, Liu et al might be a good place to turn
- some tools for downloading sras: https://github.com/saketkc/pysradb , e.g. https://colab.research.google.com/github/saketkc/pysradb/blob/master/notebooks/02.Commandline_download.ipynb
- https://github.com/ncbi/sra-tools
- a nice command for seeing cluster usage: /project/qcb_640/local/bin/clstat ( you can add an alias in your bashrc )
- place to download some kallisto indexes https://github.com/pachterlab/kallisto-transcriptome-indices
TSS Buckets / Visualization
Steps/Notes:
- new TSS buckets after RefSeqTranscript clean-up worked! Tested with a subset (~100, 1000 TSS windows)
- Figured out good visualization settings with imshow()
Next Steps:
- Put it all together, get TSSwindows for only the expressed transcripts, ordered by transript's FPKM
- Map both CTCF-TSS binding and FPKM levels together on same y-axis
11.04.2021
TSS and FPKM heatmap TOGETHER 👯
Notes/Steps:
- Building TSS buckets for RNA-seq data, ordered by delta FPKM
- Mapped TSS locations from refTranscripts onto RNAseq FPKM dataframe (annotated file from Elphege supp. excel) using the gene name
- Strange: not all gene names in RNAseq data were present in RefSeq set.
- For now, just going to clean them out (less than 2%).
- hopefully building our own DE gene RNAseq set will eliminate this issue? So that we are all using the same set of unique identifiers/references end-end of pipeline
- Again received chrom index out of bounds error
- removing chromosome X, looks like it's small enough that this issue could be coming from it? https://www.ncbi.nlm.nih.gov/grc/mouse/data
- filter out any TSS that are not within this region
- probably points to some other issue, or something I'm not understanding so we should keep note of this!
- Going back and attempting to run pybbi on full set of TSS from refseq (just to see if my matching of TSS to RNAseq is where these values become confused)
Ran full set of intervals to do bbi stackup (under header: Getting closest TSS to regions bound by CTCF)
Why are these different dimensions??
input: (refTSSwindow.size) --> 131931
output: (matrix.shape) --> (43977, 20)
11.05.2021
Download and storage for SRA files
References:
- overview: https://www.ncbi.nlm.nih.gov/sra/docs/sradownload/
- downloading sra tools to download directly for ncbi: https://github.com/ncbi/sra-tools/wiki/
- Other option is cloud delivery, might be an option to look into later GCP - google cloud
Steps
- Downloaded SRA files for Wapl set at 0h, 6h, 24h, 48h, 96h ( Wapl-C20_Xh_RNA_rep2; Mus musculus; RNA-Seq )
- ncbi group acc: SRP216929; each acc: SRX9106184, SRX9106186, SRX9106188, SRX9106190, SRX9106192
Next Steps:
- Look into package manager, options to download all SRA files (compare w
- Try kallisto!
11.10.2021
Trying Kallisto
Downloads/Prep/Resources
- In kallisto FAQs: "Transcriptome fasta files for model organisms can be downloaded from the Ensembl database. We recommend using cDNA fasta, specifically the *.cdna.all.fa.gz files. kallisto can build indices directly from gzipped files."
- Downloaded transcriptomes for multiple mouse transcriptomes into dir
/scratch/pokorny/kallisto_resources/ensembl_cdnaby navigating ensembl ftp downloads- GRCm39 is most up-to-date mus musculus genome assembly, downloaded file:
Mus_musculus.GRCm39.cdna.all.fa.gz - mm10 = GRCm38, file:
Mus_musculus.GRCm38.cdna.all.fa.gz - mm9 = NCBIM37 = GRCm37, file:
Mus_musculus.NCBIM37.65.cdna.all.fa.gz
- GRCm39 is most up-to-date mus musculus genome assembly, downloaded file:
Steps
- Submitting as jobs, and also tried on personal computer
kallistoquant4.job --> output4, from kallisto.job4 = JOBID 6629286 kallistoquant5.job --> output5, from kallisto.job5 = JOBID 6629287, activated biovenv
Results
11.11.2021
Todo:
- read paper/tutorial: https://colab.research.google.com/drive/17E4h5aAOioh5DiTo7MZg4hpL6Z_0FyWr#scrollTo=qVh9frJDgVQ-
- look into indexing / reading
- https://arxiv.org/pdf/1506.05101.pdf
- knowledge graphs
- neo4j, cypher
11.12.2021
Steps:
- Made new jupyter notebook for easier organization
- Generated Fig 6.a heatmap (did not do clustering, maybe later?)
- using bioframe overlap function, mapping RefSeq transcripts to RNAseq data
- filter by: distance, greatest amount of coverage -- choose only one refseq
Todo:
- redo stack-up, with all 4 of the auxin conditions
- remove "unchanged" RNAseqs, only include those with large deltas --> maybe should have just done that with orig. data in orig. notebook but we've come too far now
- Do same analysis with sleuth results!
11.15.2021
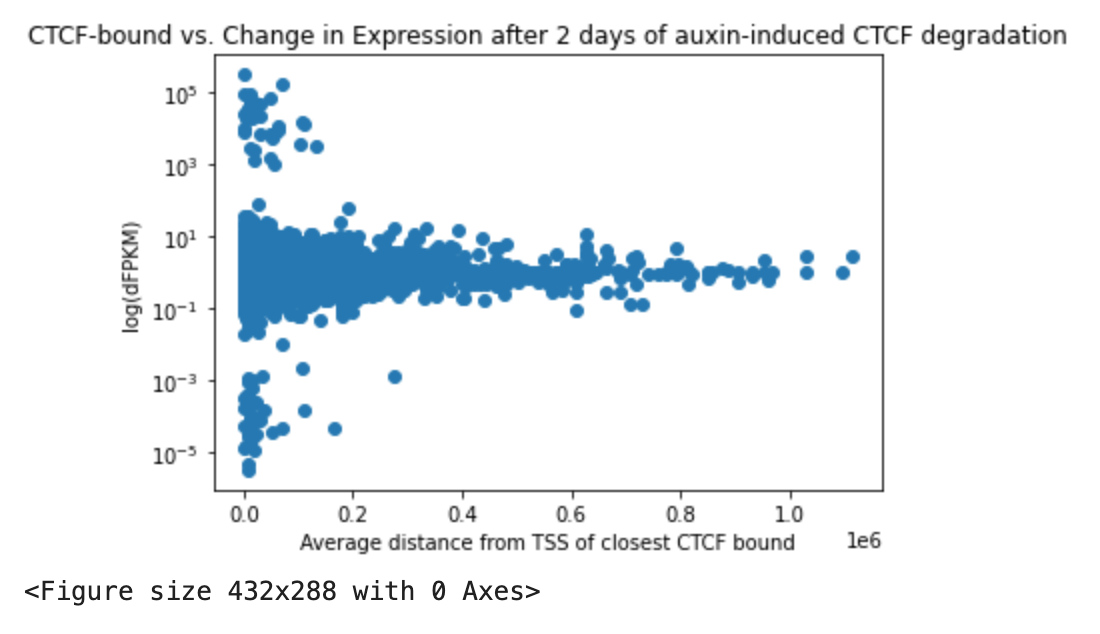
Figure 6(b) Fixed
Overview: Started a new notebook to approach figure 6, starting with mapping TSS from the beginning. The goal here is to build it more cleanly, and make functions so that can do the analysis across multiple sets of data.
Notebook Name: Elphege_2017_Fig6.
Steps/Notes:
- Noticed issues with trying to take log of delta
- At first, delta was calculated as the difference between the auxin-treated RNA-seq FPKM and the untreated). Can't take log of negative numbers.
- Also tried defining delta as (x_treated - x_original) / x_original.
- this gave negative change as very small decimals. I was trying to take log of that and it was essentially deleting all down-regulated transcripts.
- FIXED: took log10change from the beginning: log10(x_treated/x_original). Use this normalized value to compare/sort the change in expression.
- Put into a couple of
- We have figures!
Questions:
- what is n in Fig 6(b)? I wasn't able to re-do that
- cutoff=0.1 seems best to me. Is there a way to choose a cutoff other than visualization?
11.16.2021
- Finished Fig 6
- Meeting with Geoff
11.17.2021
https://learning.cyverse.org/projects/kallisto_tutorial/en/latest/step3.html
11.23.2021
Working on downloading RNA-seq files from Elphege
Purpose: Want to get RNA-seq files from Elphege_2017 to run through kallisto, sleuth pipeline and have a standard format for processing. Would be good to establish a pipeline for processing RNA-seq data for differential expression analysis.
Process:
- Trying to use package manager: pysradb
- downloaded all files from project to
/scratch/pokorny/Elphege_2017/SRP106652/pysradb_downloads/SRP106652/- Issue: this is EVERYTHING - mixture of fastq, Hi-C data, etc all with files in .sra format.
- downloaded all files from project to
Better Process:
- Use Entrez search with project number and rna-seq to download only the rna-sequencing data
- have some way to verify whether paired-end or not for kallisto input
todo:
- use SraAccList.txt to download only RNA-seq datasets
- feed all files from that into slurm job:
fastq-dump -N 7164180 -X 14328358 -O /tmp/pfd_qtpv_w_r/1 --split-files --gzip {SRR5517500.sra_files_list} - kallisto for all these output files (also in slurm job?)
- look into https://github.com/snakemake-workflows/rna-seq-kallisto-sleuth
- does this have what we need?
- maybe use this and input entire directory of .sra / fastq files. I think you just run the snakeFile in the directory with the needed files??
12.24.2021
Still downloading and running rna-seq and kallisto for Elphege et. al
- I'm downloading metadata and downloaded the list of files manually (in SraAccList.txt)
- Entrez search might be a good tool if we want to automate this --> https://www.ncbi.nlm.nih.gov/books/NBK179288/
- Todo: need to parse the 'condition' out of 'Experiment Title' column in
metadata/sra_run_summary.csv
Useful resources:
- For using loading a conda environment for carc job submissions: https://www.carc.usc.edu/user-information/user-guides/software-and-programming/anaconda
- Sleuth tutorial is a little more helpful than the one on their website: https://github.com/Kapeel/Sleuth-tutorial-iPlant
- querying ncbi API for SRA files using python: https://bootstrappers.umassmed.edu/guides/main/python_get_sra_run_ids.html
todo: