Journal Entry Assignment #2: Differential Gene expression and Preliminary ORA
The objective of this assignment is to take the normalized expression data that was created in Assignment #1 and then rank genes according to the differential expression that was performed on the normalized expression data. Furthermore, With that ranked list perform thresholded over-representation analysis(ORA) to highlight dominant genes/themes in the top set of genes for the dataset. Additionally this assignment and all the information conducted should be a standalone piece of work, as in the information and ideas from Assignment 1 will be referenced but they are not the primary focus here.
Time used: 18 h
Date started: 2022/03/10 Date completed: 2022/04/16
The activities and tasks for the assignment can be found here.
Note that the answers to these questions and requirements can be found in the matching headings of the HTML document for this assignment with a general idea and walkthrough of my thought process found below, in order of logical progression whilst working through the assignment.
Here is the link to the dataset for this assignment that the analysis was performed on; GSE193417, with the accompanying paper titled Lower Levels of GABAergic Function Markers in Corticotropin-Releasing Hormone-Expressing Neurons in the sgACC of Human Subjects With Depression of which can be found here at this link on here and its respective DOI: 10.1016/j.jbc.2022.101652.
The procedure and general skeltal structure for this assignment is as follows:
- Decide on a model design to be used for calculating differential expression
- Fit a linear model to this design, and then compute statistics of differential expression using whatever method would work best
- Sort genes by lowest p-values
- Correct p-values with multiple hypothesis correction, thereby adding a p. adj value data for each gene
- Show differentially-expressed genes with a volcano plot/MA plot
- Visualize top hits with heat map
- Run a ORA + thresholded gene set enrichment analysis
- Interpret results
I decided not to opt for making my Assignment 1 file part of my Assignment 2 file through the use of "Child nodes" because not everything I did or used in the Assignment 1 was correct or to my liking. Some things, after having learned more about data analysis and R caused me to change my perspective on the things I did. Additionally, based on feedback from both Professor Isserlin and TA Tamar Av-Shalom there were some things I needed to change and do differently. One such example is the MDS plot, which wasn't correct as it did not use the intended values. We can see in Figure 1 the MDS plot that there is clustering and therefore can safely assume that there is a strong relationship between the genes and their effects that are being investigated. This was the same conclusion that was determined in Assignment 1, even after correction, as well as using edgeR to normalize and clean the data as we had done in the previous assignment, we made some corrections and formatting changes so that the data table header was more presentable.
After downloading, cleaning, and analyzing RNASeq data, we use the Benjamini-Hochberg (BH) correction method to adjust for outliers. We also used the BH method of correction because it is less stringent, we used Benjamini-Hochberg. Bonferroni is more stringent, so we'll get fewer hits. It is preferable to use a less stringent correction because we want a reasonable number of results for our downstream analysis and do not need to worry about the cost of testing differential expression.
With this in mind, I created a volcano plot, MA plot, which has a very similar spread in terms of their data points. We can see that the genes that were expressed in a nice cluster. Then I ordered the datapoints of the top hits, i.e. for the genes as a heatmap. Those were values that were less than a threshold of p <0.05 as that seems to be the magic number.
Now the heatmeat shows the cluster of the different genes being expressed and at what concentration for each sample. We can see that there is a pattern that emerges, as mentioned in the document. Those that are MDD affected have a distinct pattern where the top half of the heatmap is primarily purple, whilst the bottom half is primarily orange and vice versa for those in the control group.
So we can see here that the authors' method to determine significant genes and perform differential gene analysis differed from my approach as we used Over Expression Analysis (ORA). I had a total of 608 genes after correction for over-representation, 280 that were upregulated, and 328 of them were downregulated.
Gene enrichment analysis is performed with g:profiler, specifically gprofiler2. The authors of the paper used false discovery rate (FDR) correction using the fdrtool package, and differential expression significance was set at a 25% FDR rate, furthermore the pathway enrichment significance was set at p < 0.05. To identify biological pathways affected by MDD, Gene Set Enrichment Analysis (GSEA) was performed using the Wald statistic-ranked gene lists and EnrichmentMap gene-set database under their default parameters.
I chose to again use a p<0.05 as the value for the threshold as it seems to have been working really well this far, and the numbers were more or less the same as the authors, signifying that despite doing things in a different manner, the similar enough results support the ideas and methods that I am using to conduct this analysis.
After creating a list of upregulated, downregulated, and all regulated genes, I plotted them using a Manhattan plot, to see the grouping and clustering from the different gene annotation databases. Furthermore, the results returned from the annotation databases made a lot of sense, seeing as how the main idea of this paper was to test how cortisol affects sgACC and the related pathways. What this means is that the databases that returned results for my analysis and the genes in question had a direct relationship. We can see that the downregulated genes offered us the most information as they had the largest amount of hits, implicating that those who have MDD have had some sort of genetic contribution and thus causes them to be more susceptible to this disease.
Now to talk about some of the issues that I had whilst doing this assignment, and some changes from the previous data set.
I decided to make sure and save all aspects and major steps of the analysis from the previous assignment for the simplicity of accessing values and information when I need it. I had some trouble getting the values and data structures to play nice. As the formats were sometimes not compatible. This was an oversight on my part as some of the data and objects I was using were not the ones I had intended or were formatted incorrectly and thus lead me to getting the wrong results. This is why downloading everything and redoing some aspects of the assignment led to a much better experience this time around. Thought it should be noted that even though one or two data structures or things did not get used, explicitly, I did use them implicitly, made for function calls, arguments and organization to be a lot better.
I did not require to do any more threshold reduction as the number of genes that I had were somewhat reasonable. Furthermore, the authors of the paper (Hyunjung et. al., 2022) had similar differentially expressed genes. The authors of the paper identified 307 genes showing significant group differences 168 upregulated, 139 downregulated and gene sets were significantly altered in those with MDD, 528 gene sets, 267 upregulated, 261 downregulated. Comparatively, we can see how close these numbers are. The large difference of 838 genes being differentially regulated and my 608 are most likely due to the fact that
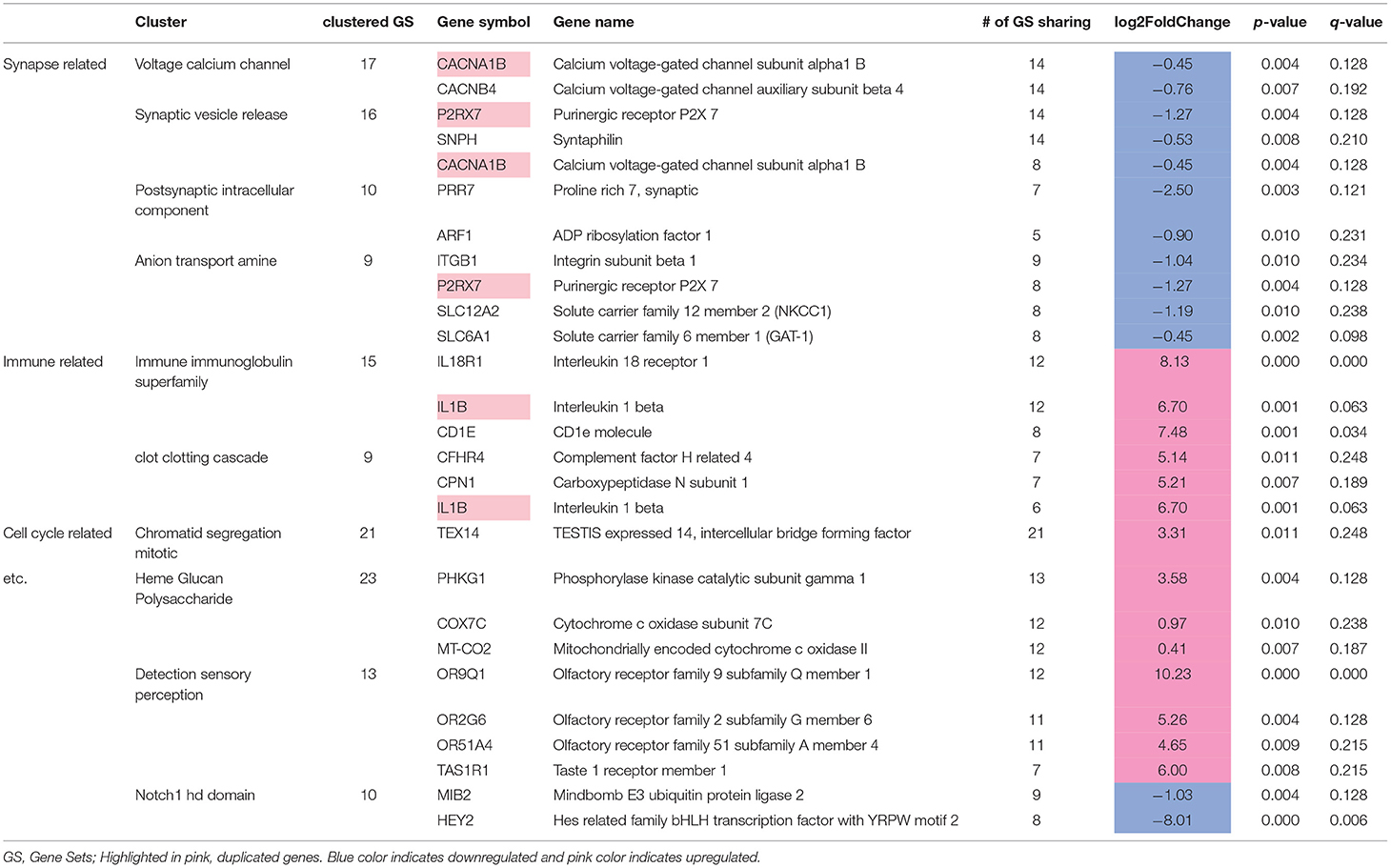
The other paper that I mentioned in my document, also dealt with and support my claims and ideas of it being a significant conclusion. But even with the research, the results the implications and background reasoning persists, as genes encode for everything in us, and if genes malfunction or there is some sort of regulatory inconsistencies, therefore causing differential gene expression to also become inconsistent we deviate from the intended functions and behaviours in not only our cells but ourselves. So, with the link between those that suffer from MDD and those that do not suffer from MDD is defintely related to the genes in this case, and as the paper suggested. For more information refer to their Table 5 which shows the 24 edge case genes and their function and their regulation status. Some of the genes on this list also coincide with mine. MT-CO1 and the 6 downregulated genes GAS6, CCDC93, METRN, TAF1, GAK, WIF1.
{kind=link}
I added the gprofiler outputs and plots as a direct query link in my document because we had nearly the same results. The only difference was the number of hits for the plots, which can be explained by the thresholded analysis methods that I was using, and the gprofilers input. The links can be found in the html document, in their respective sections.
In closing, I would like to say that more samples would have really been interesting to see, especially see if the heatmap colour pattern was more than a coincidence, as even though there are 6 samples for MDD and control, the authors themselves have mentioned that more samples could have lead to a stronger conclusion.
I would also like to see the method and use case from the authors' point of view and try to see if I can match their results by doing what they did.
1. Bonnin, S. (2022). 19.11 Volcano plots | Introduction to R. Biocorecrg.github.io. Retrieved 13 April 2022, from https://biocorecrg.github.io/CRG_RIntroduction/volcano-plots.html.
2. Cotter, D., Mackay, D., Landau, S., Kerwin, R., & Everall, I. (2001). Reduced Glial Cell Density and Neuronal Size in the Anterior Cingulate Cortex in Major Depressive Disorder. Archives Of General Psychiatry, 58(6), 545. https://doi.org/10.1001/archpsyc.58.6.545
3. Differential Expression with Limma-Voom. Ucdavis-bioinformatics-training.github.io. (2022). Retrieved 13 April 2022, from https://ucdavis-bioinformatics-training.github.io/2018-June-RNA-Seq-Workshop/thursday/DE.html.
4. Duan, E. (2022). R|Py notes: Volcano plots with ggplot2. R|Py notes. Retrieved 13 April 2022, from https://erikaduan.github.io/posts/2021-01-02-volcano-plots-with-ggplot2/.
5. Falcon, S., & Gentleman, R. (2006). Using GOstats to test gene lists for GO term association. Bioinformatics, 23(2), 257-258. https://doi.org/10.1093/bioinformatics/btl567
6. Geistlinger, L., Csaba, G., Santarelli, M., Ramos, M., Schiffer, L., Turaga, N., Law, C., Davis, S., Carey, V., Morgan, M., Zimmer, R., & Waldron, L. (2021). Toward a gold standard for benchmarking gene set enrichment analysis. Briefings in bioinformatics, 22(1), 545–556. https://doi.org/10.1093/bib/bbz158
7. Huang, D., Sherman, B., & Lempicki, R. (2008). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Research, 37(1), 1-13. https://doi.org/10.1093/nar/gkn923
8. Lin, L., & Sibille, E. (2013). Reduced brain somatostatin in mood disorders: a common pathophysiological substrate and drug target?. Frontiers In Pharmacology, 4. https://doi.org/10.3389/fphar.2013.00110
9. Oh, H., Newton, D., Lewis, D., & Sibille, E. (2022). Lower Levels of GABAergic Function Markers in Corticotropin-Releasing Hormone-Expressing Neurons in the sgACC of Human Subjects With Depression. Frontiers In Psychiatry, 13. https://doi.org/10.3389/fpsyt.2022.827972
10. Peng, R. (2022). R Programming for Data Science. Bookdown.org. Retrieved 13 April 2022, from https://bookdown.org/rdpeng/rprogdatascience/.
11. Shelton, R., Claiborne, J., Sidoryk-Wegrzynowicz, M., Reddy, R., Aschner, M., Lewis, D., & Mirnics, K. (2010). Altered expression of genes involved in inflammation and apoptosis in frontal cortex in major depression. Molecular Psychiatry, 16(7), 751-762. https://doi.org/10.1038/mp.2010.52
12. Steipe, B., & Isserlin, R. (2022). BCB420 - Computational System Biology. Bcb420-2022.github.io. Retrieved 13 April 2022, from https://bcb420-2022.github.io/General_course_prep/index.html#attributions.
13. Steipe, B., & Isserlin, R. (2022). BCB420 - Computational System Biology. Bcb420-2022.github.io. Retrieved 13 April 2022, from https://bcb420-2022.github.io/R_basics/.
14. Steipe, B., & Isserlin, R. (2022). BCB420 - Computational System Biology. Bcb420-2022.github.io. Retrieved 13 April 2022, from https://bcb420-2022.github.io/Bioinfo_Basics/.
15. Lecture modules: https://q.utoronto.ca/courses/248455/modules
16. Uku Raudvere, Liis Kolberg, Ivan Kuzmin, Tambet Arak, Priit Adler, Hedi Peterson, Jaak Vilo: g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update) Nucleic Acids Research 2019; doi:10.1093/nar/gkz369 [PDF].
The works on this page are licensed under the Creative Commons Attribution 4.0 International License, {CC BY 4.0}
Other copyright information can be found here for the course.
Placeholder for overall references. Specific references will be made explicitly.